PUBLICIDAD

Introducción: Se realiza un estudio de los nuevos fármacos antiepilépticos (lamotrigina. vigabatrina, oxcarbamazepina, topiramato etc) y su acción como medicación anticomicial y neuroprotectora.
Objetivos: Conocer la forma en que actúan los FAE en el sistema nervioso central (SNC) en el control de las epilepsias y como algunos de estos medicamento actúan como neuroprotectores, lo que es capaz de evitar o corregir las alteraciones neuropsiquiátricas que acompañan a la epilepsia.
Desarrollo: Se hace una revisión sobre los mecanismos de neuroprotección para evitar el deterioro que producen las crisis prolongadas de epilepsia. Se presenta el modo de acción de los nuevos FAEs en la fisiopatología de la epilepsia.
Conclusiones: Los nuevos FAEs pueden además de mejorar el control de las crisis de epilepsia tener un efecto favorable con las alteraciones neuropsicológicas mediante un mecanismo de neuroprotección.
Efecto neuroprotector de los nuevos fármacos antiepilépticos.
Salvador González Pal; Erick González Delgado.
Liga Cubana contra la Epilepsia
36A No 114 Apto 10 e/ 1ra y 3ra. Playa
C. P. : 11300. Ciudad de la Habana
Ciudad Habana. Cuba
Resumen
Introducción: Se realiza un estudio de los nuevos fármacos antiepilépticos (lamotrigina. vigabatrina, oxcarbamazepina, topiramato etc) y su acción como medicación anticomicial y neuroprotectora.
Objetivos: Conocer la forma en que actúan los FAE en el sistema nervioso central (SNC) en el control de las epilepsias y como algunos de estos medicamento actúan como neuroprotectores, lo que es capaz de evitar o corregir las alteraciones neuropsiquiátricas que acompañan a la epilepsia.
Desarrollo: Se hace una revisión sobre los mecanismos de neuroprotección para evitar el deterioro que producen las crisis prolongadas de epilepsia. Se presenta el modo de acción de los nuevos FAEs en la fisiopatología de la epilepsia.
Conclusiones:Los nuevos FAEs pueden además de mejorar el control de las crisis de epilepsia tener un efecto favorable con las alteraciones neuropsicológicas mediante un mecanismo de neuroprotección.
Introducción
Las crisis epilépticas han sido descritas desde hace más de 2000 años a. de n. e. , en el antiguo Egipto y en Grecia donde se hizo referencia a esta enfermedad. 1. Aunque la descripción que estas culturas hicieron sobre las crisis era bastante acertada, la brusquedad con la que se presentaban los episodios ictales y la rapidez en desaparecer acompañados con algunas manifestaciones clínicas espectaculares, hicieron que se mal interpretara la causa de estos episodios, por lo que a esta enfermedad se le denominó “mal sagrado” al creerse que la causa era debida a poderes sobrenaturales, con la participación de los dioses, de los espíritus o del demonio2.
Así el termino epilepsia deriva del griego “epilambaneim” que significa “coger por sorpresa”. Alamenon Crotona, 500 años a. n. e. señalaba que la epilepsia se originaba en el cerebro, mientras que Hipócrates, 460 años a. n. e. , en su obra “De la enfermedad Sagrada” indicaba que esta era una enfermedad, ni más divina, ni más sagrada que las demás.
El concepto de epilepsia sufrió variaciones durante el pasar de los años hasta que surgen definiciones que incluyen el verdadero origen cerebral de las crisis, así la primera definición enunciada con un concepto científico adecuado surge en la segunda mitad del siglo IXX por Hughlings Jackson que la define como: “Descarga local ocasional, rápida y excesiva de gran mal”3.
Tantos siglos de malas concepciones sobre el origen cerebral de la epilepsia repercutió en el atraso a que fueron sometidos los estudios sobre la forma de curar este mal. Así la historia de los fármacos antiepilépticos (FAE).
· En 1857 cuando John Locock (médico psiquiatra inglés) descubre en forma indirecta las propiedades antiepilépticas de los bromuros al ser utilizados en pacientes agitados con trastornos sexuales.
· En 1912 Hauptmann introduce el fenobarbital y barbitúricos en general, medicamentos que vinieron a revolucionar el tratamiento de las epilepsias.
· En 1937, Merrit y Putnan introducen la difenilhidantoina y prácticamente con estos dos medicamentos (fenobarbital y fenitoina) se manejó la mayoría de los paciente durante largos años, aunque quedaban algunos tipos de crisis sin solucionar o controlar (ausencias, mioclónicas, crisis parciales complejas y espasmos infantiles).
· En los años 60 a 70 aparecen las benzodiazepinas, la carbamazepina y el ácido valproico, terminando así el arsenal terapéutico de la epileptología.
· La década del 80 no aportó cambios, ni grandes aportes.
· En los 90 en que se vuelven a introducir en el mercado productos muy prometedores como son la lamotrigina, vigabatrina, gabapectina, topiramato, oxcarbamazepina entre otros considerados los nuevos FAE.
La lucha de los llamados nuevos fármacos antiepilépticos (FAE) es la de penetrar en la pugna con los antiepilépticos convencionales y deben cumplir algunos de los siguientes parámetros:
1) Servir para el control de la epilepsia como tratamiento único en forma de monoterapia
2) Producir bajos efectos tóxicos principalmente del SNC4 .
Objetivos
El objetivo de este trabajo es:
· El conocer la forma en que actúan los FAE en el sistema nervioso central (SNC) en el control de las epilepsia.
· Hacer una revisión sobre los nuevos mecanismos de neuroprotección para evitar el deterioro de las crisis prolongadas de epilepsia.
· Presentar una revisión de los nuevos FAEs su modo de acción en la fisiopatología de las epilepsia.
Definiciones
En primer lugar definimos los términos de crisis eléctrica, crisis epilépticas, zonas de epileptogénesis, y epilepsia :
crisis eléctrica: Es una descarga del neuronal excesiva y paroxística que puede tener una traducción clínica o no. De tener un evento ictal se denomina crisis de epilepsia (ver concepto siguiente). 5
crisis epilépticas (ictus o evento ictal): Es un paroxismo de carácter temporal producido por excesivas descargas de las neuronas de la corteza cerebral, que ocasiona un evento discernible para la persona que está experimentando la crisis o un observador. Las manifestaciones clínicas de las crisis pueden tomar diferentes formas y varían según la zona cortical afectada en el paciente por lo que reflejan las funciones de los tejidos corticales en los que se originan y propagan las excesivas descargas. 5
Zona de epileptogénesis: Es la zona neuronal que es propensa a la formación de las crisis eléctricas que llevan a crisis de tipo epilépticas. 5
Epilepsia: Es la reiteración o recurrencia de crisis de epilepsia en un mismo individuo sin causa real en el momento de producirse lleva a la epilepsia. 5
Es muy importante conocer la clasificación de las crisis de epilepsia y de las Epilepsias, ya que este es un punto importante al poner el tratamiento con FAEs.
Desarrollo
Fisiopatología y mecanismo de acción de los antiepilépticos
Vimos con anterioridad que existe una zona conocida (zona epileptogénica) que es donde comienza el proceso de la epilepsia, esta zona actúa generando diferentes cambios a nivel bio-molecular y lo produce por factores genéticos o por factores ambientales. Los cambios del nivel molecular son seguidos por el reclutamiento de poblaciones neuronales que van cambiando localmente. Existe además un período de reorganización sináptica con la acción de factores homeostásicos y factores tróficos con mitosis desorganizada. Una despolarización desordenada y aumentada.
Las descargas excesivas producen crisis de epilepsia y excitotoxicidad. Las que pueden producir más daño neuronal, más despolarización y más crisis de epilepsia, de tal manera se establece un circulo vicioso negativo en el que mientras más frecuentes se produzcan las crisis de epilepsia, habrá mayor daño neuronal, por lo que de no interrumpirse el proceso epiléptico se pueden llegar a ver cambios degenerativo irreversibles de la neurona.
El intercambio iónico en la zona de epileptogénesis:
El potencial de la membrana en reposo oscila entre – 70 a – 90 mV, cuando se activa y despolariza para llevar a cabo sus funciones, puede este potencial llegar a +30 mV o producir también despolarizaciones parciales (en las dendritas) con cambios que pueden bajar entre -40 y -50 mV, estos cambios de valores están en dependencia del estímulo. Cuando las neuronas se hacen más negativas con otro tipo de estímulo se consideran que están hiperpolarizadas.
En el potencial de reposo existe un equilibrio iónico, calcio (Ca), sodio (Na) y cloro (Cl) fuera de la membrana y potasio (K) con proteinas ionizadas en el interior. El potencial de reposo depende fundamentalmente del potasio. La membrana neuronal es, en reposo, solamente permeable al potasio.
En la despolarización neuronal normal hay varios mecanismos:
a) entrada de sodio a la célula.
b) Entrada de calcio a la célula
c) Entrada de cloro a la célula
d) Salida de potasio
El potencial de acción es dependiente del sodio.
Los neurotransmisores y su acción sobre el potencial de membrana.
El cerebro usa la glucosa como fuente de energía primaria, el ácido de glutámico, o glutamato es un metabolito común de metabolismo de glucosa (vía el ciclo de Krebs); por lo que, el ácido glutámico está presente en todo el tejido del cerebro, y en el fluido de extracelular del cerebro.
Figura 1
Acción del glutamato.
El glutamato está envuelto en varios procesos metabólicos en el cerebro:
· En la síntesis de proteínas y péptidos.
· Precursor para el neurotransmisor inhibitorio, ácido gamma-amino butírico (GABA).
La observación de niveles elevados de glutamato en el fluido extracerebral, asociado al aumento de la actividad del cerebro hace que este sea considerado como un neurotransmisor excitador, sin embargo, aunque la toxicidad excitatoria inducida por el glutamato es el mecanismo más frecuente que conduce al daño neuronal otras explicaciones necesitan ser realizadas. 7
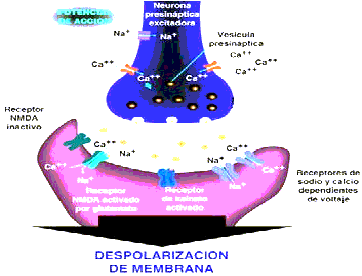
En las neuronas localizadas en el foco epileptogénico que sufren fenómenos de despolarización de membrana, con aparición de potenciales de acción repetitivos y sostenidos, tanto durante la crisis como en el intervalo interictal. En la génesis de tales fenómenos interviene activamente glutamato, que interactúa con varios tipos de receptores postsinápticos. De estos el más importante es el NMDA (N-metil-D-Aspartato), que sirve como compuerta para la entrada de iones como sodio y calcio, los cuales contribuyen a la despolarización sostenida de la membrana celular (Figura 1)
Las neuronas tienen tipos diferentes de receptores del glutamato en su superficie algunos de los cuales puede ser útiles para tratar las enfermedades neurológicas. La caracterización de estos receptores es compleja y continuada. Los receptores de glutamato pueden ser divididos en “Ionotropicos” y “Metabotrópicos”. 8
receptores Ionotrópicos.
La activación de receptores ionotrópicos glutaminergicos producen mayor permeabilidad de la membrana celular a los cationes de sodio (Na+) y calcio (Ca++). Hay tres tipos de receptores glutamato - Ionotrópicos, los que son distinguidos por su respuesta a el AMPA (alfa-amino-3-hidroxi-5-metilo-isoxazoleproprionato), al acido kainitinico, o al NMDA (N-metilo-D-aspártico).
El AMPA y los receptores de kainato son entidades distintas y separadas, aunque bastantes similares químicamente, tienen una estimulación individual difícil y muchos problemas de estimulación cruzada. Estos receptores de glutamato están compuestos por varias subunidades de proteínas. Cuatro subunidades del receptor glutamato (GluR1-GluR4) sirven como receptor de la subunidad AMPA , y cinco sub-unidades del mismo receptor constituyen la sub-unidades del receptor de kainato (GluR5-GluR7 y KA1-KA2). El receptor completo esta formado por una combinación de estas subunidades. El papel de receptores kainato se conoce pobremente, pero se ha sugerido que la activación de este produce la entrada de Ca++.
Los receptores AMPA de glutamato son permeables al Na+ (tienen efecto excitador) y parecen ser relativamente impermeables a Ca++ esto se cumple principalmente en el estado "normal" de la neurona. La sub-unidad de GluR2, una vez unida a otra sub-unidad de GluR, forma canales que son relativamente impermeables a la entrada de Ca++ y una sub-unidad GluR2 de baja regulación que permite la formación de canales que aumentan el flujo de Ca++ lo que puede llevar a la muerte celular ("hipótesis de GluR2"). 9. 10
Los receptores de NMDA están compuestos de 5 subunidades que pertenecen a dos familias--NMDAR1 y NMDAR2. La familia de NMDAR2 tiene cuatro miembros--A, B, C, y D. El receptor de NMDA es voltaje-dependiente, y usa el magnesio (Mg++) como regulador del canal. Bajo condiciones asociadas con la despolarización de la membrana, el Mg++ se cambia de sitio y permite que cantidades relativamente grandes de calcio entren a la célula. Con la entrada del calcio comienza una cadena de reacciones bioquímicas dentro de la neurona que pueden conducirla a su muerte. El calcio es descrito como el mediador primario de toxicidad excitatoria que produce daño neuronal11.
Se continua estudiando el papel fisiopatológico de los receptores de NMDA por su aparente involucración en la etiología de daño del cerebral en el curso de las crisis de epilepsia, la isquemia, el trauma craneoencefálico y las enfermedades neurodegenerativas. Se ha realizado un esfuerzo para desarrollar fármacos que bloqueen los receptores de NMDA. Algunas de estos han sido llamados bloqueadores de receptores de NMDA y se han comprobados en animales y humanos.
Desgraciadamente, muchos (como MK801) han demostrado tener efectos adversos inaceptables, que han restringido su aplicación. Esto pudiera estar relacionado en que el receptor NMDA no sólo está envuelto en los procesos de aprendizaje y memoria, si no también en la muerte celular (bajo las circunstancias extremas). Sin embargo, los agentes que actúan sobre las subunidades NR2B del receptor de NMDA pueden tolerarse mejor, por lo que el bloqueo de los receptores NMDA es una área activa de investigación12.
Receptores metabotrópicos:
Los receptores de Metabotropicos son una familia de receptores del glutamato que actúan indirectamente en los canales iónicos a través de un segundo sistema de mensajero. Las subunidades de los receptores Metabotrópicos de glutamato se describen con un sistema de anotación similar al de los receptores Ianotropicos de glutamato, sólo que la letra "m" aparece delante, como en el mGluR3.
Como indica la tabla 3, los agonistas de estás subunidades metatrópicas inducen las convulsiones, mientras los antagonistas de otros subtipos tienen las propiedades del anticonvulsivantes. Estas observaciones aumentan considerablemente la complejidad de la investigación en esta área.
Tabla 1. SUBTIPOS DE receptores METABOTRÓPICOS GLUTÁMICOS Y SU rol EN LA EPILEPSIA
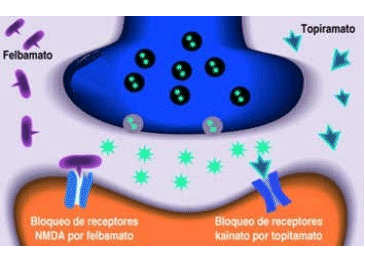
Una forma de inhibir las descargas epileptiformes consiste en bloquear los receptores para glutamato, mediante compuestos como felbamato (nuevo FAE), que tiene actividad antagonista sobre los receptores NMDA. Tal parece que topiramato, un nuevo agente anticonvulsivo de reciente desarrollo también actúa a este nivel al bloquear los receptores de tipo kainato5, 14
Figura 2 El bloqueo selectivo de los receptores glutaminérgicos, ya sea aquellos NMDA o los de tipo kainato, es una estrategia efectiva para el control de la epilepsia, pues de este modo se inhibe la generación de potenciales postsinápticos excitadores.
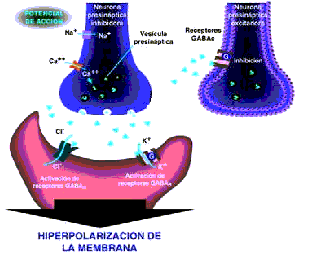
Figura 3. Generación de potenciales postsinápticos inhibidores. El neurotransmisor GABA activa dos tipos diferentes de receptores produciendo entrada de cloro y salida de potasio lo cual hiperpolariza la membrana haciéndola menos excitable.
Otra alternativa para impedir la génesis y propagación de las descargas neuronales consiste en hiperpolarizar la neurona mediante la activación de receptores tipo A para ácido gamma aminobutírico o GABA (receptores GABAA) proceso que permite el paso de iones de cloro al interior de la célula, disminuye el potencial de reposo y hace que la membrana sea menos excitable. Un efecto similar se obtiene al estimular los receptores GABA tipo B (GABAB), los cuales están ligados a proteínas G de membrana y al ser activados, indirectamente producen la apertura de los canales para potasio. Estos mecanismos generan potenciales postsinápticos inhibidores que se oponen a los potenciales postsinápticos excitadores, mediados por glutamato. En la epilepsia los mecanismos anteriormente explicados están alterados lo que facilita la persistencia y diseminación de las descargas epileptiformes.
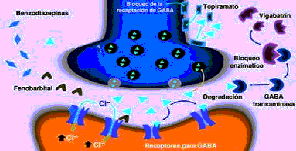
Para controlar las crisis epilépticas, otra de las vías sería el favorecer la acción de los mecanismos inhibidores, mediados por ácido gamma aminobutírico (GABA), entre los medicamentos que favorecen la acción del GABA por activación de sus receptores (GABAA) se encuentran las benzodiacepinas y los barbitúricos.
Otra estrategia fármaco – terapéutica consiste en aumentar los niveles disponibles de GABA en la terminal sináptica. El vigabatrín es un FAE que bloquea la enzima GABA transaminasa (encargada de degradar dicho neurotransmisor) y la tiagabine actúa al inhibir los procesos de recaptación a nivel de la terminal presináptica y aumenta de esta forma la disponibilidad de GABA en la sinapsis. Finalmente, aunque no se conoce con exactitud el mecanismo antiepileptogénico del ácido valproico, topiramato y gabapentín, las pruebas disponibles indican que aparentemente están relacionados con el incremento en el funcionamiento de las sinapsis gabaérgicas
Figura 4:Control de la epilepsia mediado por mecanismos dependientes de GABA. Las benzodiacepinas, barbitúricos y nuevos antiepilépticos como vigabatrín y tiagabine favorecen la actividad gabaérgica, aumentando la inhibición sobre las neuronas que generan las descargas paroxísticas.
Los canales de sodio son indispensables para la generación y propagación de los potenciales de acción, tanto en epilepsia como en condiciones fisiológicas esto los hacen ser de gran importancia para el control de las crisis de epilepsia, de esta forma los FAE como la fenitoína, carbamacepina, ácido valproico, felbamato y lamotrigine actúan sobre ellos. La mayoría de estos agentes son útiles para el control tanto de las crisis parciales como de las generalizadas, ya que actúan mediante un bloqueo selectivo de los canales rápidos de sodio, mecanismo muy efectivo para el control de la epilepsia.
A nivel del tálamo existen corrientes de calcio de bajo umbral, exclusivas de esta región del cerebro y conocidas como corrientes T. Estas intervienen en los procesos de sincronización tálamocortical, que acompañan a cierto tipo de epilepsias generalizadas y por consiguiente, constituyen un fundamento terapéutico muy interesante. De hecho, en la actualidad, numerosos investigadores han orientados sus esfuerzos a identificar sustancias que actúen a este nivel, aunque hasta el momento solo se ha comprobado que la etosuximida y ácido valproico son los únicos dos fármacos con capacidad reconocida para modular la actividad de las corrientes T.
Por último se trabaja en alternativas terapéuticas que actúen sobre los canales de potasio de los receptores neuropeptídicos y de los receptores de adenosina.
Fármacos candidatos para la Neuroprotección en la Epilepsia: Las drogas antiepilépticas (FAEs).
Los mecanismos de neuroprotección se pueden explorar mediante la potenciación de la actividad protectora de los FAEs con cualidades químicas bien conocidas. El daño causado por las crisis de epilepsia inducidas en animales de experimentación ha sido estudiada. Además, este procedimiento nos permite apreciar y comparar el efecto de los FAEs en dos vertientes diferentes, el antiepiléptico y el neuroprotector, este último mediante el esclarecimiento de los mecanismos que llevan al daño neuronal.
Existe consenso en que los cambios en los niveles de Ca++ intracelular conduce a un daño neuronal. Un ligero incremento de los niveles de Ca++ intercelular es necesario para la transmisión sináptica y para numerosas funciones metabólicas intracelulares. Por lo que, elevaciones pequeñas y transitorias del Ca++ intercelular ocurren en la actividad neuronal normal. Sin embargo, las elevaciones moderadamente prolongadas de Ca++, como las que pueden ocurrir en el curso de algunas crisis de epilepsia prolongadas o de un estado de mal epiléptico (más de una hora) conducen a la muerte de la célula neuronal. 15
Protección Neuronal. 15Si recordamos los conceptos de crisis eléctrica, epilepsia, y epileptogénesis enunciados con anterioridad vemos que en la zona de epileptogénesis se producen cambios fisiopatológicos que además de desencadenar los episodios ictales pueden mediante la acumulación del Ca++ activar un proceso neurodegenerativo.
La neuroprotección no es más que la prevención de que no ocurra una descompenzación de la zona epileptogénica mantenida con las consecuentes crisis de epilepsia a repetición, esto se puede conseguir mediante diferentes mecanismos:
a) Fármacos capaces de prevenir la acumulación en exceso del de Ca++ intercelular.
b) Fármacos que evitan los procesos que activan la necrosis o apoptosis que siguen la acumulación persistente de Ca++ .
c) Fármacos que eviten o prevengan los cambios compensatorios como son la gliosis, la reorganización de la sinapsis o la neo-neurogénesis.
No es sorprendentemente, que entre los candidatos más prometedores para protección nerviosa contra las crisis inductoras del daño de neuronal está los FAEs. Así los FAEs no solo son de utilidad ante el control de las crisis de epilepsia sino que nos permiten abordar la neuroprotección mediante otros mecanismos no relacionados al efecto anticonvulsivante. El mecanismo de muchos otros fármacos se evalúa en la actualidad en el contexto de daño isquémico del cerebro pero en nuestra discusión se basa el daño neuronal relacionado con las crisis de epilepsia y no con el observado en las isquemias.
Mecanismos de Acción de FAEs
Los mecanismos de acción de FAEs es tema de intensa investigación. Se han enunciado muchos y diferentes mecanismos de acción de estos medicamentos y se ha comprobado que estas acciones son las responsable de su eficacia clínica en la epilepsia, pero como planteamos con anterioridad otros mecanismos de acción de los FAEs menos estudiados es el de su uso como agentes neuroprotectores.
Si comprendemos adecuadamente fisiopatología de la epilepsia y la de la muerte neuronal, se hace factible comprobar que hay fármacos con doble efecto: antiepiléptico y neuroprotector para evitar la muerte cerebral. Aunque por el momento no está claro que las propiedades de los FAEs útiles en el control de las crisis sean las mismas que puedan servir en la neuroprotección.
Los FAEs actualmente disponibles son un grupo heterogéneo de productos químicos con propiedades parecidas pero con una sorprendente banda de efectos biológicos. Como una forma de simplificar este tema agrupamos las drogas según el efecto bloqueador de los canales y mecanismos envueltos en el daño neuronal. En general, como fue explicado con anterioridad los FAEs actúan de la siguiente forma:
· inhibición de la activación repetitiva y sostenida de los canales de Na+ (bloqueo de canales)
· Perfeccionamiento de inhibición mediada a través de GABA.
· Atenuación de actividad de canales de Ca++ voltaje-dependientes
· Disminución en la excitación mediada por el glutamato
Tabla 4. FAEs INHIBICIÓN DE LOS CANALES DE NA+ Y ESTABILIZACIÓN DE LAS MEMBRANAS NEURONALES.
Como se ha visto en la tabla 4, diversos FAEs intervienen y actúan en el bloqueo de los canales de Na+, aunque hasta el momento esto es lo más demostrado su actividad continua en estudio. El efecto de inhibición de los canales de Na+ interfiere con la propagación de los potenciales de acción y disminuye la liberación de neurotransmisores, entre los que se incluye el neurotransmisor excitador. 5, 14, 16
tabla 5. FAEs QUE ACTÚAN SOBRE CANALES DE VOLTAJES SENSITIVOS AL CA++
El calcio desempeña numerosos papeles en el funcionamiento neuronal. Mediante el bloqueo de los canales de Ca++ se pueden producir numerosos efectos. :
1 - El bloqueo de los canales de calcio tipo T es el tratamiento eficaz en las crisis de ausencia de pequeño mal.
2 - El bloqueo de los canales de Ca++ de alto voltaje (tipo- L) esta asociado con el control de crisis parciales con o sin generalización secundaria.
3 - La liberación del neurotransmisor es otra consecuencia del bloqueo de los canales de Ca++ lo que produce una descarga reducida de neurotransmisores, (entre los que se incluye al glutamato).
4 - Elevados niveles de Ca++ intercelular activan numerosos procesos Ca++ dependientes que llevan a la muerte celular. El bloqueo de los canales de Ca++ puede jugar un papel central previniendo estas progresiones.
En la tabla 5 vimos los FAEs que actúan en los diferentes tipos de canales de Ca++.
Tabla 6. FAEs CON EFECTOS ANTAGÓNICOS PARA LA EXCITACIÓN MEDIADA POR glutamato.
La mayoría de los FAEs disponible no han mostrado actuar directamente sobre los receptores de glutamato. Como la tabla 6 indica, sólo el felbamato ha demostrado ser eficaz en la activación del receptor de NMDA mientras que el fenobarbital y topiramato parece tener efectos antagónicos en los receptores de AMPA / Kainato
Tabla 7. FAES QUE SE RELACIONAN CON LA INHIBICIÓN A TRAVÉS DEL GABA
El ácido gamma-amino butinico (GABA) es un neurotramisor aminoacídico (el receptor GABA es parte de un complejo canal de cloruro) y la actividad de la neurona GABAergica es una fuente mayor de inhibición neuronal de la actividad basal. Las benzodiazepinas tienen un receptor único en el canal de Cloruro / GABA que produce un efecto agonista. Existen otros fármacos antiepilépticos que refuerzan o imitan la transmisión de GABA (Tabla 7) teniendo como efecto el restablecimiento de los niveles normales de inhibición, mientras que ayudan al sistema neuronal a volver a su equilibrio normal excitación-inhibición.
Se cree que los mecanismos que refuerzan la inhibición a través del GABA incluyen la síntesis reforzada de GABA y disminuyen recaptación de este. Existen algunos FAEs que se unen a otros sitios del receptor GABA y regulan la función de este por un mecanismo, de modulación alosterica del receptor. Las investigaciones actuales han indicado un papel importante del agonismo del GABA en la neuroprotección actuando en contra de lesiones inducidas por crisis, mediante su capacidad de contrarrestar la actividad de las crisis agudas continuas. Un ejemplo de esto son los barbitúricos (fenobarbital etc. ) los que se ha demostrado experimentalmente, que reducen la intensidad de reorganización de la sinapsis del hipocampo durante las crisis epilépticas en ratas adultas. 16
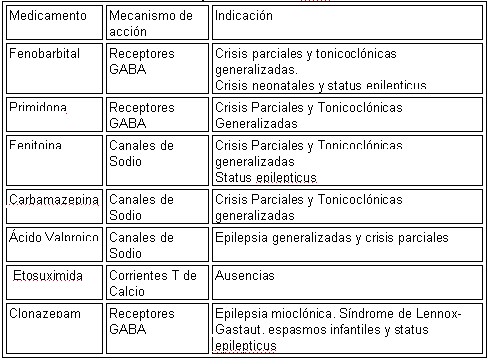
Fármacos Clásicos y Nuevos para el control de la epilepsia 17
El objetivo del tratamiento farmacológico de la epilepsia consiste en reducir al máximo la frecuencia e intensidad de las crisis, con el mínimo de efectos colaterales. Para esto se cuenta con medicamentos como fenobarbital, primidona, carbamacepina, fenitoína, etosuximida, clonazepam y ácido valproico, los cuales han constituido por décadas las únicas alternativas disponibles en el arsenal terapéutico neurológico. El mecanismo de acción e indicaciones de tales fármacos ha sido bien estudiado y existen recomendaciones claras acerca de sus usos terapéuticos.
Fármacos convencionales.
En la tabla 8 podemos ver las recomendaciones acerca del uso de los fármacos antiepilépticos convencionales.
Tabla 8. RECOMENDACIONES SOBRE EL USO DE LOS FAEs CLÁSICOS.
Es así que el medicamento de elección para el manejo de los pacientes con crisis generalizadas tónicoclónicas es ácido valproico, mientras que carbamacepina y fenitoína son considerados como fármacos de segunda línea. En individuos con crisis mioclónicas o ausencias, las recomendaciones actuales favorecen el uso, en primera instancia, de ácido valproico, reservando otros medicamentos como clonazepam para los casos que no responden a éste.
En las crisis parciales con o sin generalización secundaria es preferible iniciar con fenitoína o carbamacepina, aunque ácido valproico también constituye una alternativa eficaz. A pesar que fenobarbital y primidona son eficaces en el tratamiento de las crisis parciales, mioclónicas y tónicoclónicas generalizadas, la severidad de los efectos colaterales, sobre todo somnolencia y trastornos del aprendizaje, han limitado su uso sin olvidar la posibilidad de adicción que produce.
Nuevos fármacos antiepilépticos
Aunque el objetivo ideal en el tratamiento de la epilepsia consiste en controlar la enfermedad por completo y sin efectos farmacológicos adversos, esta meta no ha sido alcanzada. De hecho, entre 25% y 30% de los pacientes son refractarios a los esquemas tradicionales. No obstante, con la introducción al mercado de nuevos antiepilépticos, muchos de los enfermos anteriormente intratables pueden ser controlados. Los compuestos descubiertos en años recientes incluyen felbamato, oxcarbacepina, lamotrigine, vigabatrín, clobazam, gabapentín, topiramato, tiagabine, zonisamida y stiripentol. Además, se espera que en pocos años el número de éstos aumente, ofreciendo mejores alternativas para los diferentes tipos de epilepsia 5, 14, 17, 19
En 1996 fue introducida al mercado oxcarbacepina, un análogo de carbamacepina con igual mecanismo de acción y las mismas indicaciones de esta última. Su ventaja principal es que posee un mejor perfil farmacocinético y ocasiona menos efectos colaterales, de modo que es una alternativa especialmente atractiva para el control de los pacientes con crisis parciales. También en el mismo año fue comercializada fosfenitoína, un compuesto similar a fenitoína, pero para administración parenteral. Tiene dos ventajas sobre su antecesor, por una parte, la posibilidad de administración intramuscular y por otra, una menor incidencia de efectos adversos.
Felbamato: 19
La primera de las nuevas drogas antiepilépticas, fue autorizado su uso en los EEUU en 1993 como monoterapia y como terapia simultanea para el tratamiento de crisis parciales y crisis tonicoclónicas secundarias. Esta también demostró gran eficacia en el tratamiento de síndrome de Lennox-Gastaut siendo aprobada para estas indicaciones.
Estudios clínicos documentan su eficiencia contra un amplio espectro de crisis primarias generalizadas, incluidas epilepsia mioclónica juvenil, típica y atípica ausencias, espasmos infantiles y crisis primarias tonicoclónicas.
El felbamato posee un gran número de acciones celulares. Ha sido reportado que aumenta los niveles de activación GABA, disminuye la activación de NMDA, limita los potenciales de acción de alto voltaje mediante el bloqueo de los canales de sodio y aumenta la neuroprotección contra la hipoxia (in Vitro). Basado en esta multiplicidad de acción el felbamato tiene un gran espectrun en actividad antiepiléptica, además de algunos efectos secundarios favorables para ciertos pacientes como sedante y perdida de peso.
Gabapectina: 19
Este fármaco posee múltiples acciones celulares como, el aumento de los niveles cerebrales de GABA y su liberación, bloqueo de lo canales L de calcio. Posee limitada acción a través de mecanismos desconocidos que incrementan la serotonina libre en el plasma y brinda gran neuroprotección contra la toxicidad neuronal producida por el glutamato (in Vitro).
La gabapectina es un acticonvulsivante de estrecho espectrum usado particularmente en el control de crisis parciales, secundarias y generalizadas, y primarias tonicoclónicas; no ha sido encontrado que este sea activo en contra de otros tipos de crisis primarias generalizadas, y ausencias generalizadas.
Su licencia de uso fue dada de EEUU en 1993, y la seguridad de su tolerabilidad son las bases de su utilidad clínica, este no es metabolizado o fusionado con proteínas, es excretado de forma intacta por la orina, y no posee significativa interacción con otros fármacos.
Conclusiones
Primero que todo es de gran importancia el conocimiento del el diagnostico de la epilepsia, su clasificación con el propósito de tratar solamente al paciente que lo necesita y utilizar la clasificación de las crisis y del las Epilepsias para escoger el tratamiento idóneo con FAEs.
En este trabajo se ha abordado los diferentes mecanismos y formas en que actúan los fármacos antiepilépticos y se ha visto que las vías fundamentales son:
inhibición de la activación repetitiva y sostenida de los canales de Na+ (bloqueo de canales)
inhibición GABA-mediada
Atenuación de la actividad de los canales de Ca++ voltaje-dependientes
Disminución en la excitación mediada por el glutamato
Se ha visto como desde 1912 se comenzaron a proponer medicamentos como fenobarbital, Primidona, Fenitoina, carbamacepina, Ácido Valproico, Etosuximida, Clonazepam y muchos otros que cumplían con estos requisitos y que por décadas fueron la única opción de neurólogos para el tratamiento de las epilepsias.
Se expuso además como a partir del 1996 han surgido en el mercado nuevos fármacos como Vigabatrin, Tiagabina, topiramato, lamotrigina, Gabapectina, Felbamato, entre otros que vinculan el control de las crisis epilépticas con la neuroprotección.
Recomendaciones
1. Ampliar el conocimiento de la enfermedad para realizar el diagnóstico adecuado de epilepsia.
2. Conocer las Clasificaciones de las crisis de epilepsia.
3. Adecuar el tratamiento con Fármacos antiepilépticos según la clasificación de las crisis.
4. Conocimiento del proceso de degeneración neuronal que se puede ver en los pacientes con crisis de epilepsia.
5. Utilización de FAEs que sean además de tener su efecto anti- ictal actúen como neuroprotectores.
Bibliografía
1. KinnierJV, Reynolds EH. Translation and analysis of a cuneiform text forming part of a Babylonian treatise on epilepsy. Medici Histori. 1990; 34:185-98.
2. Inglis B. Historia de la Medicina. Barcelona, Eds. Gribalbo, 1968
3. Bellomo, LE. La Historia de la enfermedad Epiléptica según Owsei Temkin. Neuropsiquiqatría. 1976; 7: 2 Julio Diciembre: 26 – 35.
4. Vitieri Torres C. Avances en el tratamiento farmacológico de las epilepsias. I Congreso Iberoamericano Virtual de neurología. neurologia. rediris. es/congreso-1/conferencias/epilepsia-6. html.
5. Guberman Alan and Bruni J. Capitulo 1 Terminology: In Essential of Clinical Epilepsy. Butterworth Heinemann 1999
6. Commision on classification and terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. epilepsia 1989; 30: 389 – 399.
7. Choi DW. Excitotoxic cell death. J Neurobiol. 1992; 23:1261-1276
8. Moshé L. Solomon, MD and Decker F. John, PhD. Neuroprotection and Epilepsy. Neurology treatment update. pharmacotherapy. medscape. com/Medscape/Neurology/TreatmentUpdate/2000/tu01/public/toc-tu01. html
9. Friedman LK, Pellegrini-Giampietro DE, Sperber EF, Bennett MV, Moshe SL, Zukin RS. Kainate-induced status epilepticus alters glutamate and GABAA receptor gene expression in adult rat hippocampus: an in situ hybridization study. J Neurosci. 1994; 14(5 Pt 1):2697-2707
10. Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997; 20:464-470.
11. Sutula T, Cascino G, Cavazos J, Paranda I. Mossy fiber synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988; 239:1147-1150
12. Rogawski MA. Neuroprotection: Implications for the future clinical management of epilepsy, a satellite symposium of the 53rd Annual Meeting of the American Epilepsy Society; Orlando, Fla; Dec. 5, 1999
13. Merlin L. From presentation made at an Investigator's Workshop: Glutamate metabotropic receptors as a therapeutic target in epilepsy. Annual Meeting of the American Epilepsy Society; Orlando, Fla; Dec. 5, 1999
14. Sander J. W. and Hart Y. M. : Epilepsy questions and answers. Merit Publishing International. 1999
15. DeLorenzo RJ. Neuroprotection: Implications for the future clinical management of epilepsy, a satellite symposium of the 53rd Annual Meeting of the American Epilepsy Society; Orlando, Fla; Dec. 5, 1999.
16. Eriksson a. Knutsson E. and Nergardh. The effect of Lamotrigine on Epileptiform Discharges in Young Patiens with Drug-Resistant Epilepsy Epilepsi, 42, (2); 230-236, 2001
17. Shank P. Richard, Gardocki F. Joseph, Streeter J. Anthony and Maryanoff E. Bruce. An Overview of the preclinical aspects of topiramate: Pharmacology, Pharmacokinetics, and Mechanism of Action epilepsia, 41(Suppl. ):S3-S9, 2000.
18. Sutula T, Cavazos J, Golarai G. Alteration of long-lasting structural and functional effects of kainic acid in the hippocampus by brief treatment with phenobarbitol. J Neurosci. 1992; 12:4173-4187.
19. McLean Michael. Management of convulsive disorders with th newer antiepileptic grugs: Current review: In Broadening the spectrum of clinical usus of antiepileptic drugs. The Royal Society of Medicine Press Limited. 1999.
20. Hardus P. , Verduin M. W. , Engelsman M. , Edelbroek M. P. , Segers, P. J. , Brendschot M. J. T. T. and Stilma S. J. Visual field loss associated with Vigabatrin: Quantificatión and relation to dosage. epilepsia 42(2), 262-267, 2001.
IMPORTANTE: Algunos textos de esta ficha pueden haber sido generados partir de PDf original, puede sufrir variaciones de maquetación/interlineado, y omitir imágenes/tablas.





")
La importancia de la adherencia al tratamiento en la epilepsia
Adamed Laboratorios
Fecha Publicación: 10/12/2024
Abordaje psiquiátrico del paciente epiléptico
Patricia Serrano de la Fuente et. al
Fecha Publicación: 20/05/2024
Depresión en la epilepsia
Carmelo Pelegrín Valero
Fecha Publicación: 18/05/2023
Epilepsia con Psicosis
Nora Ines Muros Cobos et. al
Fecha Publicación: 20/05/2022
")
Epilepsia conceptos
Eddy Monge
Fecha Publicación: 19/06/2021
Normalización forzada, a propósito de un caso
Sandra Arilla Andrés et. al
Fecha Publicación: 21/05/2021