PUBLICIDAD

Trastorno de ansiedad generalizada: Etiopatogenia.
Pilar Alejandra Sáiz Martínez.
Profesora Titular de psicología Médica
Factores biológicos
Estudios genéticos
Hasta la fecha no existen pruebas fehacientes de la implicación de factores genéticos en la etiología del trastorno de ansiedad generalizada (TAG). El estudio realizado por Noyes et al (1987), pone de manifiesto que los familiares de pacientes con TAG tienen un mayor riesgo de padecer el trastorno que los familiares de sujetos control. No obstante, dicho riesgo no fue superior al riesgo de padecer TAG entre familiares de pacientes con trastorno de pánico. Por otra parte, dos trabajos que utilizan criterios de inclusión DSM-III (Torgersen, 1983; Andrews et al, 1990), realizados en Noruega y Australia, respectivamente, refieren tasas de concordancia similares para el TAG entre gemelos monocigotos y dicigotos. Dos estudios posteriores (Kendler et al, 1992; Skre et al, 1993), que utilizan criterios de inclusión DSM-IIIR, constatan que la heredabilidad del TAG se relaciona con la de la depresión y, sugieren, que ambos trastornos podrían compartir factores genéticos similares. Por último, otro estudio realizado recientemente (Chantarujikapong et al, 2001), que incluye a más de 3300 parejas de gemelos de sexo masculino, pone de manifiesto que TAG, trastorno de pánico (TP) y trastorno de estrés postraumático (TEPT) comparten factores genéticos comunes, de modo que el riesgo de que se produzca sintomatología de TAG es debida a un 37. 9% de contribución genética común a síntomas de TP y TEPT.
Los estudios de genética molecular son aún muy escasos, no obstante, se ha descrito la posible asociación entre un polimorfismo del gen del transportador de serotonina, que daría lugar a una disminución de la recaptación de la misma por parte de la célula presináptica, y determinados rasgos de ansiedad en pacientes y sus familiares (Lesch et al, 1996).
En definitiva, puede decirse que la causalidad genética del TAG está en entredicho, no existiendo una evidencia clara de que dicho trastorno se transmita de modo independiente. De modo que los factores ambientales podrían tener gran relevancia en su génesis (Nurnberger y Berretini, 1998).
Neurobiología
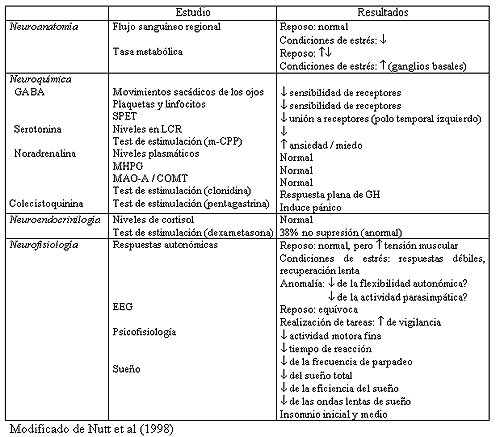
En la tabla 1 se resumen los principales hallazgos
realizados en esta área en relación al TAG.
Tabla 1 Estudios neubiológicos en el TAG
. Neuroanatomía
Flujo sanguíneo regional cerebral
En los estudios de neuroimagen no se constatan, en situación de reposo, diferencias en el flujo sanguíneo regional o global entre pacientes con TAG y sus correspondientes controles. Sin embargo, en los pacientes con TAG, ante situaciones de estrés psicológico, se detecta un descenso del flujo cerebral en la mayoría de las regiones cerebrales (Mathew et al, 1982).
Metabolismo regional cerebral
Hasta la fecha se han realizado varios estudios, que utilizando tomografía de emisión de positrones (TEP), miden el metabolismo de la glucosa como indicador de la actividad cerebral. Así, Wu et al (1991), refieren que durante la realización de tareas pasivas los pacientes con TAG presentan una mayor tasa metabólica que los controles en algunas áreas cerebrales como determinadas zonas de los lóbulos occipital, temporal y frontal, en el cerebelo, y en el tálamo. De igual modo, se ha descrito tanto un incremento (Buchsbaum et al, 1987), como un decremento de actividad en los ganglios basales (Wu et al, 1997).
Durante la realización de tareas activas se observa un incremento relativo de la tasa metabólica en ganglios basales (Wu et al, 1991). Por otra parte, el tratamiento con benzodiacepinas genera un decremento en el metabolismo del cortex occipital, del sistema límbico y de los ganglios basales (Buchsbaum et al, 1987).
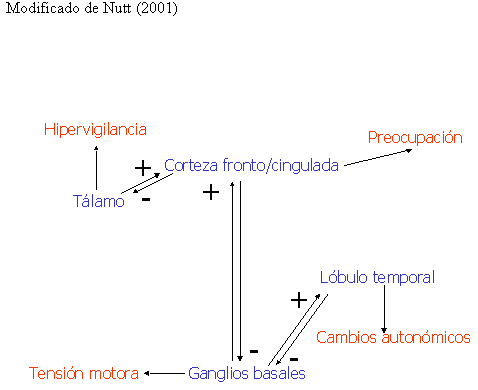
Los escasos hallazgos existentes parecen sugerir la existencia de circuitos cerebrales hiperactivos en el TAG. En la figura 1 se resumen los circuitos cerebrales posiblemente implicados en el TAG, así como, su relación con la sintomatología de este trastorno.
Figura 1 Circuitos cerebrales y síntomas en el TAG
. Neuroquímica
Acido Gamma-aminobutírico (GABA)
El GABA es el principal neurotransmisor inhibidor del sistema nervioso central. El papel de las benzodiacepinas (BZ) como agentes ansiolíticos ha traído como consecuencia el interés en el estudio del sistema GABA/BZ en el TAG. Se ha hipotetizado que los pacientes con TAG podrían tener una deficiencia de dicho sistema, bien debido a una reducción de la sensibilidad receptorial, posiblemente debido a una reducción del número de receptores, o bien debido al déficit de neurotransmisores endógenos inhibidores (Nutt, 2001).
En pacientes con TAG se ha descrito la existencia de un menor número de lugares de unión de BZ en plaquetas (Weizman et al, 1987) y linfocitos (Ferrarese et al, 1990). Existiendo, de igual modo, dos estudios que ponen de manifiesto que el tratamiento con BZ produce un incremento del número de lugares de unión periféricos (Weizman et al, 1987; Rocca et al, 1991).
La medición de la velocidad de los movimientos sacádicos de los ojos constituye un modo adecuado de evaluación de la sensibilidad de los receptores benzodiacepínicos centrales. Utilizando este modelo, existen estudios (Cowley et al, 1993; Kroboth et al, 1998), que sugieren una hiposensibilidad de dichos receptores en el TAG.
Por otra parte, el estudio mediante SPECT de la distribución de los receptores benzodiacepínicos cerebrales pone de manifiesto que los pacientes con TAG presentan, respecto a los controles, una mayor homogeneidad en la distribución de dichos receptores, así como, un decremento de los mismos en el lóbulo temporal izquierdo (Tiihonen et al, 1997). Los autores concluyen que esos hallazgos son acordes con la hipótesis de que una heterogeneicidad en la perfusión, metabolismo y densidad de receptores es necesaria para mantener la adaptabilidad del organismo y, dicha capacidad, está ausente en los estados de ansiedad.
Noradrenalina (NA)
El complejo locus coeruleus-noradrenalina-sistema nervioso simpático desempeña un papel crítico en la respuesta ante situaciones de alarma y miedo. Numerosos investigadores han tratado de desvelar la existencia de posibles anomalías del sistema catecolaminérgico en el TAG.
Existen estudios que no encuentran diferencias, entre pacientes con TAG y controles, en los niveles plasmáticos de NA (Mathew et al, 1982), ni en los niveles de los enzimas responsables de la degradación de NA (catecol orto-metiltransferasa, dopamina b-hidroxilasa y monoamino oxidasa) (Khan et al, 1986). No obstante, un estudio más reciente (Kelly y Cooper, 1998), pone de manifiesto que los pacientes con TAG tienen niveles plasmáticos de NA superiores a los de los controles. Aunque dichas diferencias no alcanzaban una significación estadística podrían sugerir cierta hiperactividad noradrenérgica en el TAG.
La realización de una estimulación con clonidina (agonista alfa2-adrenérgico) da lugar, en pacientes con TAG, a respuestas planas de la hormona de crecimiento, mientras que en sujetos control estimula su liberación (Abelson et al, 1991). No obstante, se desconoce si esa respuesta anómala detectada en pacientes con TAG es debida a una regulación a la baja de los receptores a2 (consecuencia de la hiperactividad crónica del sistema noradrenérgico central que tienen los pacientes con TAG) o, sin embargo, es debida a la existencia de mecanismos de retroalimentación anómalos para la hormona de crecimiento (Abelson et al, 1991).
La clonidina es capaz, por otra parte, de suprimir el sueño REM sin causar una disrupción del sueño. Schittecatte et al (1995), estudian el efecto de la administración de clonidina en la latencia REM en pacientes con TAG, depresión mayor, depresión menor y un grupo control. En condiciones basales el tiempo de latencia REM1-REM2 se situó en torno a los 100 minutos en todos los grupos estudiados. Sin embargo, tras la administración de clonidina, el intervalo REM1-REM2 se eleva a, aproximadamente, 300 minutos tanto en los controles como en los pacientes con TAG y depresión menor, mientras que en los pacientes con depresión mayor ese incremento es mínimo. Este hallazgo parece sugerir que en los pacientes con TAG existe una sensibilidad normal de los receptores a2 (Nutt, 2001).
La administración de yohimbina (antagonista presináptico alfa2-adrenérgico) da lugar a una respuesta atenuada de 3-metoxi-4-hidroxifenilglicol en pacientes con TAG respecto a los controles (Charney et al, 1989), lo que sugeriría una disminución de la sensibilidad de los receptores alfa2-presinápticos.
Por último, citar que existen estudios que ponen de manifiesto la existencia de un menor número de lugares de unión a2-adrenérgicos en plaquetas de pacientes con TAG (Sevy et al, 1989; Cameron et al, 1990).
A pesar de posibles discrepancias, tomados en conjunto, los datos existentes podrían sugerir la existencia de una hiposensibilidad de los receptores noradrenérgicos en pacientes con TAG. Dicha hiposensibilidad podría ser consecuencia de una adaptación a largo plazo frente a una hiperactividad noradrenérgica crónica (Connor y Davidson, 1998; Nutt, 2001).
Serotonina (5-HT)
La 5-HT está implicada en la mediación de la ansiedad a través de las vías que se originan en los núcleos del rafe y que inervan, entre otras áreas cerebrales, el sistema límbico, el hipotálamo y el tálamo. Hasta la fecha se han descrito varios receptores 5-HT pre y postsinápticos. Al menos dos de ellos (5-HT-1A y 5-HT-2) han sido implicados en el TAG.
El efecto beneficioso de la buspirona (un agonista parcial 5-HT1A) en el TAG, puede explicarse a partir del hecho de que la activación de los receptores presinapticos 5-HT1A da lugar a una reducción inicial en la liberación de 5-HT, que se sigue de un incremento de la transmisión serotoninérgica.
Por otra parte, la estimulación de los receptores postsinápticos 5-HT-2 da lugar a conductas de evitación y ansiedad (Graeff, 1991). En este sentido, se ha comprobado los potenciales efectos ansiolíticos de las sustancias con acción antagonista 5-HT-2, tales como la nefazadona, trazadona o mirtazapina.
Otras evidencias de la implicación de la 5-HT en el TAG derivan de la comprobación de que los pacientes con TAG tienen menores niveles de 5-HT en LCR que los sujetos control (Brewerton et al, 1995). Además, en los pacientes con TAG se ha detectado un menor número de lugares de unión para paroxetina en plaquetas (Iny et al, 1994). De igual modo, la administración de m-clorofenilpiperacina (un agonista no específico 5-HT-1 y 5-HT-2), provoca un incremento de ansiedad y hostilidad en pacientes con TAG (Germine et al, 1992). Por último, recordar el ya mencionado posible papel de un polimorfismo del gen del transportador de serotonina en la génesis de determinados rasgos de ansiedad (Lesch et al, 1996).
En resumen, los hallazgos existentes parecen demostrar un anormal funcionamiento del sistema serotoninérgico en el TAG. Los hallazgos existentes sugieren, de igual modo, que los fármacos con efectos reguladores del sistema serotoninérgico (por ej. , agonistas 5-HT-1A, 5-HT-2 antagonistas o inhibidores selectivos de recaptación de serotonina) podrían tener un efecto beneficioso en este trastorno.
Colecistoquinina (CCK)
La CCK es un péptido abundante en el sistema nervioso central, que interactúa con otros neurotransmisores como el GABA, la NA y la 5-HT. La evidencia clínica de una posible implicación de este neurotransmisor en el TAG procede de un estudio en el que la inyección intravenosa de pentagastrina (un agonista del receptor tipo B de la CCK), es capaz de provocar ataques pánico en un mayor porcentaje de pacientes con TAG que de controles (Brawman-Mintzer et al, 1997). Sin embargo, la utilización de antagonistas de la CCK en pacientes con TAG ha generado resultados pocos esperanzadores (Adams et al, 1995). Por tanto, puede decirse que el papel de la CCK en el TAG es, en la actualidad, incierto.
Factor liberador de corticotropina (CRF)
El CRF está presente en numerosas regiones cerebrales relacionadas con las respuestas de ansiedad y miedo (amígdala, locus coeruleus, complejo dorsal vagal) (Butler y Nemeroff, 1990). Sin embargo, hasta la fecha no existen datos clínicos que apoyen la implicación del CRF en el TAG, ya que en un estudio realizado por Fossey et al (1996), no se encuentran diferencias en los niveles de CRF entre pacientes con TAG y el grupo control.
. Neuroendocrinología
Eje hipotálamo-hipófiso-adrenal (HHA)
La activación del eje HHA es un importante componente de la respuesta normal ante situaciones de estrés. Kelly y Cooper (1998) refieren niveles plasmáticos de cortisol normales en pacientes con GAD.
Una prueba muy utilizada para evaluar el funcionamiento del eje HHA es la prueba de supresión con dexametasona: la realización de esta prueba en pacientes con TAG (Avery et al, 1985; Tiller et al, 1988), pone de manifiesto unas tasas de no-supresión del 38% y 27%, respectivamente, sugiriendo la existencia de posibles anomalías en la regulación del HHA en este trastorno.
Neuroesteroides
Recientemente se ha puesto de manifiesto que parte de los efectos beneficiosos de los antidepresivos en el TAG podrían derivarse del incremento que producen de los efectos moduladores de los neuroesteroides sobre el GABA (Romeo et al, 1998). No obstante, para conocer la verdadera relevancia de los neuroesteroides en el TAG son necesarios más estudios.
Neurofisiología
Función autonómica
Hoehn-Saric et al (1989), refieren que los pacientes con TAG presentan ante situaciones de estrés una menor actividad electrodérmica (evaluada a través de la conductancia de la piel), frecuencia respiratoria, presión arterial y variabilidad de la frecuencia cardíaca, que los pacientes controles. Sin embargo, dichas diferencias no se observan en situación de reposo. Estos resultados indicarían la existencia de una inhibición (en vez de un incremento) simpática en los pacientes con TAG (Nutt et al, 1998). Por su parte, Cameron et al (1990), refieren una menor presión arterial sistólica, en situación de reposo, en los pacientes con TAG que en controles. Así mismo, Thayer et al (1996) refieren una menor variabilidad de la frecuencia cardíaca en pacientes con TAG.
Los resultados de esos estudios sugieren que el TAG se asocia a una disminución de la flexibilidad autonómica, posiblemente debido a una reducción del tono vagal (Connor y Davidson, 1998).
Hoehn-Saric et al (1997), refieren que los pacientes con TAG presentan, en situación de reposo, un incremento de la tensión muscular respecto a los controles. Ese incremento de la tensión muscular podría constituir un mejor reflejo de la mayor activación del sistema nervioso central que otras medidas fisiológicas periféricas (Nutt et al, 1998).
Electroencefalograma (EEG)
Los datos existentes en situación de reposo son equívocos. Se ha sugerido que la actividad hemisférica derecha, que se supone relacionada con el procesamiento de emociones negativas, podría estar intensamente relacionada con el incremento del tono muscular periférico, facilitando de este modo los comportamientos de fuga asociados a los estados de ansiedad (Hoehn-Saric et al, 1997). La realización de EEG durante las últimas horas de la mañana pone de manifiesto la existencia de una hipervigilancia en los pacientes con TAG (Saletu-Zyhlarz et al, 1997).
Psicofisiología
En el estudio mencionado con anterioridad (Saletu-Zyhlarz et al, 1997) se demuestra, así mismo, que los pacientes con TAG tienen una disminución de la actividad motora fina, una disminución de la respuesta de parpadeo y un deterioro del tiempo de reacción.
Sueño
El insomnio es un síntoma frecuente en el TAG, de modo que más del 70% de estos pacientes refieren problemas de sueño.
En los pacientes con TAG se evidencia una disminución del tiempo total de sueño (Saletu-Zyhlarz et al, 1997), una peor eficiencia del mismo (Saletu-Zyhlarz et al, 1997), una mayor latencia de sueño (insomnio de conciliación) y mayor frecuencia de fracturas de sueño (insomnio de mantenimiento) (Reynolds et al, 1983). Sin embargo, la latencia de sueño REM y el porcentaje de sueño REM respecto al tiempo total de sueño son normales (Reynolds et al, 1983).
Resumen
Lo expuesto con anterioridad pone de manifiesto que el conocimiento de las bases neurobiológicas del TAG, es aún incipiente. No obstante, en la mayoría de evaluaciones realizadas en situación basal (reposo), los pacientes con TAG no difieren de los controles normales, aunque, sin embargo su respuesta a estresores es anómala. De modo que esas anomalías incluyen, entre otros, a los sistemas de neurotransmisión gabaérgico, noradrenérgico, serotoninérgico, eje HHA y algunas respuestas psicofisiológicas (Nutt et al, 1998).
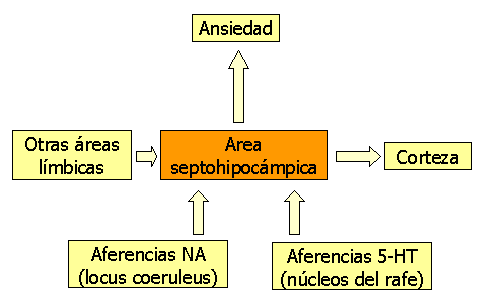
Un modo de integrar los hallazgos anteriormente descritos es mediante el Sistema Inhibidor de la conducta de Gray (1991) (figura 2)
Figura 2: modelo neuroanatómico de Gray en el TAG (Sistema Inhibidor de la Conducta) Modificado de Nutt et al (1998)
Se ha hipotetizado que dicho sistema se activa ante determinadas señales (estímulos aversivos, novedosos o ausencia de la recompensa esperada), produciendo una inhibición de la conducta en marcha (llegando al extremo de la paralización completa), un aumento del análisis sensorial del medio (dirigiendo la atención preferentemente al estímulo nuevo y potencialmente peligroso) y una potenciación de la conducta de defensa resultante. Se trataría de una respuesta que mimetizaría los estados de ansiedad continua, tal y como ocurre en el TAG (Brawman-Mintzer y Lydiard, 1997).
La base neuroanatómica de este sistema estaría integrada por el circuito de Papez (formado a su vez, por el área septal, la formación hipocámpica, los cuerpos mamilares, el tálamo anteroventral y el cortex cingulado) junto con el cortex prefrontal y las vías ascendentes noradrenérgicas (sobre todo, el fascículo dorsal que sube desde el locus coeruleus), dopaminérgicas y serotoninérgicas que inervan estas estructuras del proencéfalo (Díez y Sánchez, 2000).
Las aferencias noradrenérgicas y serotoninérgicas hacia el área septohipocámpica son fundamentales para la activación de este sistema. Concretamente, Gray propone que la inhibición de movimiento inducida por el miedo y las respuestas comportamentales ante castigo estarían mediados por el sistema serotoninérgico, mientras que las respuestas comportamentales ante la ausencia de recompensa estaría mediada por el sistema noradrenérgico.
Factores psicosociales
Relaciones familiares
En adultos, se ha descrito una asociación entre elevados niveles de ansiedad y antecedentes de bajo nivel de cuidados por parte de los padres y de sobreprotección paterna (Parker 1979, 1986; Silove et al, 1991). Concretamente, las personas con TAG refieren, frecuentemente, relaciones con los padres aberrantes, incluyendo una ausencia de cariño, y padres sobreprotectores pero negligentes (Nutt et al, 1998).
Estos hallazgos son consistentes con los referidos por Angst y Vollrath (1991), que, en su estudio de seguimiento a largo plazo, encuentran que las personas con depresión o ansiedad (TAG o trastorno de pánico), o ambas patologías, refieren más frecuentemente que los controles antecedentes de relaciones conflictivas entre los padres, conflictos con los progenitores, falta de atención o cuidados por parte de los mismos y traumas sexuales.
Acontecimientos vitales
Diversos estudios ponen de manifiesto que los acontecimientos vitales pueden tener importancia en la etiología del TAG. Así, los pacientes con TAG refieren una pérdida parental precoz más frecuentemente que los pacientes con trastorno de pánico (Torgersen, 1986). Por otra parte, el TAG es más frecuente en personas que han sufrido experiencias estresantes o traumáticas (Blazer et al, 1987; Roemer et al, 1996/97). Steketee y Foa (1987), ponen de manifiesto que el TAG es una secuela frecuente en sujetos que han estado sometidos a acontecimientos vitales específicos tales como violación o participación en combate. Brown et al (1996), concluyen que es frecuente encontrar en los 6 meses previos al desarrollo, tanto de trastornos depresivos como de trastornos de ansiedad (incluyendo el TAG), la existencia de un acontecimiento vital grave. De igual modo, se ha demostrado que en los trastornos depresivos son más frecuentes los acontecimientos relacionados con pérdida, mientras que en los trastornos de ansiedad la asociación es más intensa con acontecimientos peligrosos o amenazadores.
Es posible que los acontecimientos vitales, en particular los asociados con daño, tengan un papel causal en el desarrollo de algunos de los aspectos psicopatólogicos centrales del TAG, tales como ansiedad anticipatoria crónica y constante percepción amenaza. Además, una vez establecido el TAG, los acontecimientos traumáticos podrían facilitar el mantenimiento de elevados niveles de ansiedad (Finlay-Jones, 1989).
Modelo cognitivo
Beck y Emery (1985) proponen que la ansiedad patológica deriva de una percepción errónea del peligro. La inexactitud se genera por medio de la atención selectiva a los detalles negativos del entorno, de las distorsiones en el procesamiento de la información, y a la desmesurada percepción negativa de la persona de sus capacidades de afrontamiento.
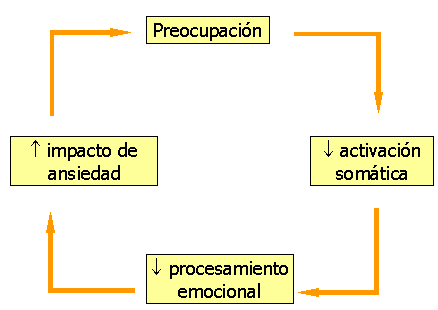
Las cogniciones ansiosas interactuarían con otros procesos manteniendo la espiral de la ansiedad (figura 3).
Figura 3: Espiral cognitiva de la ansiedad. Modificado de Nutt et al (1998)
La preocupación reduciría la reactividad somática, impididiendo un adecuado procesamiento emocional (Roemer y Borkovec, 1993), lo cual contribuiría, a su vez, al mantenimiento de las respuestas y sensaciones de ansiedad (Borkovec, 1994). Por último, las personas con TAG refieren una falta de control sobre los estímulos aversivos, lo cual incrementaría las propiedades aversivas de dichos estímulos (Harvey y Rapee, 1995).
Otras evidencia que corroboraría este punto de vista sería la utilidad de las terapias cognitivas y cognitivo-conductuales en el tratamiento del TAG (Durham y Allan, 1993).
Conclusiones
A pesar de los esfuerzos realizados hasta la fecha, es necesaria una mayor investigación que sirva para un mayor esclarecimiento de las bases etiopatogénicas del TAG. Los modelos y teorías propuestos para explicar la etiopatogenia del TAG describen a menudo fases parciales de un mismo proceso o los mismos fenómenos desde diferentes puntos de vista. Los distintos mecanismos por los que se puede provocar TAG (biológicos y psicológicos) no son excluyentes, sino que probablemente representan diferentes etapas de un proceso psicofisiológico único y complicado que puede fallar a distintos niveles.
Bibliografía
Abelson JL, Glitz D, Cameron OG, Lee MA, Bronzo M, Curtis GC. Blunted growth hormone response to clonidine in patients with generalized anxiety disorder. Arch gen Psychiatry 1991; 48: 157-162.
Adams JB, Pyke RE, Costa J, Cutler NR, Schweizer E, Wilcox CS et al. A double-blind, placebo-controlled study of a CCK-B receptor antagonist, CI-988, in patients with generalized anxiety disorder. J Clin Psychopharmacol 1995; 15: 428-43
Andrews G, Stewart G, Allen R, Henderson AS. The genetics of six neurotic disorders: a twin study. J Affect Disord 1990; 19: 23-29.
Angst J, Vollrath M. The natural history of anxiety disorders. Acta Psychiatr Scand 1991; 84: 446-452.
Avery DH, Osgood TB, Ishiki DM, Wilson LG, Kenny M, Dunner DL. The DST in psychiatric outpatients with generalized anxiety disorder, panic disorder, or primary affective disorder. Am J Psychiatry 1985; 142: 844-848.
Beck AT, Emery G. Anxiety disorders and phobia: a cognitive perspective. New York: Basic Books, 1985.
Blazer D, Hughes D, George LK. Stressful life events and the onset of a generalized anxiety syndrome. Am J Psychiatry 1987; 144: 1178-1183.
Borkovec TD. The nature, functions and origins of worry. En: Davey GCL, Tallis F eds. Worrying: perspectives in theory, assessment and treatment. England: John Wiley & Sons, 199
Brawman-Mintzer O, Lydiard RB. Biological basis of generalized anxiety disorder. J Clin Psychiatry 1997; 58 (Suppl 3): 16-25.
Brewerton TD, Lydiard RB, Johnson MR, Ballenger JC, Fossey MD, Roberts JE. CSF serotonin: diagnostic and seasonal differences. En: New research and abstracts of the 148th meeting of the American Psychiatric Association. Washington, DC: American Psychiatric Press, 1995; abstract NR358: 151.
Brown GW, Harris TO, Eales MJ. Social factors and comorbidity of depressive and anxiety disorders. Br J Psychiatry 1996; 168 (Suppl 30): 50-57.
Buchsbaum MS, Wu J, Haier R, Hazlett E, Ball R, Katz M. Positron emission tomography assessment of effects of benzodiazepines on regional glucose metabolic rate in patients with anxiety disorder. Life Sci 1987; 40: 2393-2400.
Butler PD, Nemeroff CB. Corticotropin-releasing factor as a possible cause of comorbidity in anxiety and depressive disorders. En: Maser CR, Cloninger CR ed. Comorbidity of mood disorders. Washington, DC: American Psychiatric Press, 1990; pp. 413-435.
Cameron OG, Smith CB, Lee MA, Hollingsworth PJ, Hill EM, Curtis GC. Adrenergic status in anxiety disorders: platelet alpha 2-adrenergic receptor binding, blood pressure, pulse, and plasma catecholamines in panic and generalized anxiety disorder patients and in normal subjects. Biol Psychiatry 1990; 28: 3-20.
Chantarujikapong SI, Scherrer JF, Xian H, Eisen SA, Lyons MJ, Goldberg J et al. A twin study of generalized anxiety disorder symptoms, panic disorder symptoms and post-traumatic stress disorder in men. Psychiatry Res 2001; 103: 133-145.
Charney DS, Woods SW, Heninger GR. Noradrenergic function in generalized anxiety disorder: effects of yohimbine in healthy subjects and patients with generalized anxiety disorder. Psychiatry Res 1989; 27: 173-182.
Connor KM, Davidson JRT. Generalized anxiety disorder: neurobiological and pharmacotherapeutic perspectives. Biol Psychiatry 1998; 44: 1286-129
Cowley DS, Roy-Birne PP, Greenblatt DJ, Hommer DW. Personality and benzodiazepine sensitivity in anxious patients and control subjects. Psychiatry Res 1993; 47: 151-162.
Díez C, Sánchez Planell L. Etiopatogenia. En: Vallejo J, Gastó C eds. Trastornos afectivos: ansiedad y depresión. 2ª ed. Barcelona: Masson, 2000; pp. 51-100.
Durham RC, Allan T. Psychological treatment of generalised anxiety disorder. A review of the clinical significance of results in outcome studies since 1980. Br J Psychiatry 1993; 163: 19-26.
Ferrarese C, Appolonio I, Frigo M, Perego M, Piolti R, Trabucchi M et al. Decreased density of benzodiazepine receptors in lymphocytes of anxious patients: reversal after chronic diazepam treatment. Acta Psychiatr Scand 1990; 82: 169-173.
Finlay-Jones R. Anxiety. En: Brown GW, Harris TO eds. Life events and illness. Vol I. New York: Guilford Press, 1989; pp. 95-112.
Fossey MD, Lydiard RB, Ballenger JC, Laraia MT, Bissette G, Nemeroff CB.
Cerebrospinal fluid corticotropin-releasing factor concentrations in patients with anxiety disorders and normal comparison subjects. Biol Psychiatry 1996; 39: 703-707.
Germine M, Goddard AW, Woods SW, Charney DS, Heninger GR. Anger and anxiety responses to m-chlorophenylpiperazine in generalized anxiety disorder. Biol Psychiatry 1992; 32: 457-461.
Graeff FG. Neurotransmitters in the dorsal periaqueductal grey and animal models of panic anxiety. En: Briley M, File S eds. New concepts in anxiety. London: MacMillan Press, 1991.
Gray JA. The psychobiology of fear and stress. New York: Cambridge University Press, 1991.
Harvey AG, Rapee RM. Cognitive-behavior therapy for generalized anxiety disorder. Psychiatr Clin North Am 1995; 18: 859-870.
Hoehn-Saric R, McLeod DR, Zimmerli WD. Somatic manifestations in women with generalized anxiety disorder. Psychophysiological responses to psychological stress. Arch gen Psychiatry 1989; 46: 1113-1119.
Hoehn-Saric R, Hazlett RL, Pourmotabbed T, McLeod DR. Does muscle tension reflect arousal? Relationship between electromyographic and electroencephalographic recordings. Psychiatry Res 1997; 71: 49-55.
Iny LJ, Pecknold J, Suranyi-Cadotte BE, Bernier B, Luthe L, Nair NP et al. Studies of a neurochemical link between depression, anxiety, and stress from [3H]imipramine and [3H]paroxetine binding on human platelets. Biol Psychiatry 1994; 36: 281-291.
Kelly CB, Cooper SJ. Differences in variability in plasma noradrenaline between depressive and anxiety disorders. J Psychopharmacol 1998; 12: 161-167.
Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. Major depression and generalized anxiety disorder. Same genes, (partly) different environments?
Arch gen Psychiatry 1992; 49: 716-722.
Khan A, Lee E, Dager S, Hyde T, Raisys V, Avery D et al. Platelet MAO-B activity in anxiety and depression. Biol Psychiatry 1986; 21: 847-849.
Kroboth PD, Folan MM, Bauer KS, Tullock W, Wright CE, Sweeney JA. Do alprazolam-induced changes in saccadic eye movement and psychomotor function follow the same time course? J Clin Pharmacol 1998; 38: 337-346.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996; 274: 1527-1531.
Mathew RJ, Weinman ML, Claghorn JL. Anxiety and cerebral blood flow. En: Mathew RJ ed. The biology of anxiety. New York: Brunner/Mazel, 1982; pp: 23-33.
Mathew RJ, Ho BT, Taylor D. Dopamine-beta-hydroxylase response to epinephrine injection in anxious patients and normals. Biol Psychiatry 1982; 17: 393-397.
Noyes R, Clarkson C, Crowe RR, Yates WR, McChesney CM. A family study of generalized anxiety disorder. Am J Psychiatry 1987; 144: 1019-102
Nurnberger JI Jr, Berretini W. Psychiatric genetics. London: Chapman & Hall, 1998.
Nutt D, Argyropoulus S, Forshall S. Generalized anxiety disorder: diagnosis, treatment and its relationship to other anxiety disorders. London: Martin Dunitz, 1998.
Nutt DJ. Neurobiological mechanisms in generalized anxiety disorder. J Clin Psychiatry 2001; 62 (Suppl 11): 22-27.
Parker G. Reported parental characteristics in relation to trait depression and anxiety levels in a non clinical group. Aust N Z J Psychiatry 1979; 13: 260-26
Parker G. Validating an experimental measure of parental style: the use of a twin sample. Acta Psychiatr Scand 1986; 73: 22-27.
Reynolds CF 3rd, Shaw DH, Newton TF, Coble PA, Kupfer DJ. EEG sleep in outpatients with generalized anxiety: a preliminary comparison with depressed outpatients. Psychiatry Res 1983; 8: 81-89.
Rocca P, Ferrero P, Gualerzi A, Zanalda E, Maina G, Bergamasco B et al. Peripheral-type benzodiazepine receptors in anxiety disorders. Acta Psychiatr Scand 1991; 84: 537-54
Roemer L, Borkovec TD. Worry: unwanted cognitive experience that controls unwanted somatic experience. En: Wegner DM, Pennebaker J eds. Handbook of mental control. Englewood Cliffs, NJ: Prentice Hall, 1993.
Roemer L, Molina S, Litz BT, Borkovec TD. Preliminary investigation of the role of previous exposure to potentially traumatizing events in generalized anxiety disorder. Depress Anxiety 1996-97; 4: 134-138.
Romeo E, Strohle A, Spalletta G, di Michele F, Hermann B, Holsboer F et al. Effects of antidepressant treatment on neuroactive steroids in major depression. Am J Psychiatry 1998 ; 155: 910-913.
Saletu-Zyhlarz G, Saletu B, Anderer P, Brandstatter N, Frey R, Gruber G et al. Nonorganic insomnia in generalized anxiety disorder. 1. Controlled studies on sleep, awakening and daytime vigilance utilizing polysomnography and EEG mapping. Neuropsychobiology 1997; 36: 117-129.
Schittecatte M, Garcia-Valentin J, Charles G, Machowski R, Pena-Othaitz MJ, Mendlewicz J et al. Efficacy of the 'clonidine REM suppression test (CREST)' to separate patients with major depression from controls; a comparison with three currently proposed biological markers of depression. J Affect Disord 1995; 33: 151-157.
Sevy S, Papadimitriou GN, Surmont DW, Goldman S, Mendlewicz J. Noradrenergic function in generalized anxiety disorder, major depressive disorder, and healthy subjects. Biol Psychiatry 1989; 25: 141-152.
Silove D, Parker G, Hadzi-Pavlovic D, Manicavasagar V, Blaszczynski A. Parental representations of patients with panic disorder and generalised anxiety disorder. Br J Psychiatry 1991; 159: 835-841.
Skre I, Onstad S, Torgersen S, Lygren S, Kringlen E. A twin study of DSM-III-R anxiety disorders. Acta Psychiatr Scand 1993; 88: 85-92.
Steketee G, Foa EB. Rape victims: post traumatic stress responses and their treatment: a review of the literature. J Anx Disorders 1987; 1: 69-86.
Thayer JF, Friedman BH, Borkovec TD. Autonomic characteristics of generalized anxiety disorder and worry. Biol Psychiatry 1996; 39: 255-266.
Tiihonen J, Kuikka J, Rasanen P, Lepola U, Koponen H, Liuska A et al. Cerebral benzodiazepine receptor binding and distribution in generalized anxiety disorder: a fractal analysis. Mol Psychiatry 1997; 2: 463-471.
Tiller JW, Biddle N, Maguire KP, Davies BM. The dexamethasone suppression test and plasma dexamethasone in generalized anxiety disorder. Biol Psychiatry 1988; 23: 261-270.
Torgersen S. Genetic factors in anxiety disorders. Arch gen Psychiatry 1983; 40: 1085-1089.
Torgersen S. Childhood and family characteristics in panic and generalized anxiety disorders. Am J Psychiatry 1986; 143: 630-632.
Weizman R, Tanne Z, Granek M, Karp L, Golomb M, Tyano S et al. Peripheral benzodiazepine binding sites on platelet membranes are increased during diazepam treatment of anxious patients. Eur J Pharmacol 1987; 138: 289-292.
Wu JC, Buchsbaum MS, Hershey TG, Hazlett E, Sicotte N, Johnson JC. PET in generalized anxiety disorder. Biol Psychiatry 1991; 29: 1181-1199.
Wu JC, Maguire G, Riley G, Lee A, Keator D, Tang C et al. Increased dopamine activity associated with stuttering. Neuroreport 1997; 8: 767-770.
IMPORTANTE: Algunos textos de esta ficha pueden haber sido generados partir de PDf original, puede sufrir variaciones de maquetación/interlineado, y omitir imágenes/tablas.

Delírium (I): Aspectos histórico-conceptuales, nosología, epidemiología, etiopatogenia y clínica.
Antonio Soto Loza
Fecha Publicación: 01/12/2012

Delírium (I): Aspectos histórico-conceptuales, nosología, epidemiología, etiopatogenia y clínica.
Francisco Domínguez Belloso et. al
Fecha Publicación: 03/02/2012
Trastorno por estrés postraumático. Una visión global.
Jorge González Fernández
Fecha Publicación: 01/03/2006
Etiopatogenia y tratamiento del trastorno de angustia.
Andrés Herrán
Fecha Publicación: 01/01/2004
Depresión y abuso de sustancias: hipótesis etiopatogénicas del trastorno dual.
B. Martín
Fecha Publicación: 01/01/2002
Aproximación histórica a la psicopatología de los trastornos de la conducta alimentaria.
Olga Martín
Fecha Publicación: 01/01/2000