PUBLICIDAD

Esta revisión estudia los diferentes mecanismos de acción antidepresiva, considerando los cambios bioquímicos implicados en la depresión y como éstos son modificados por el tratamiento antidepresivo, actuando más allá del nivel de la interacción entre neurotransmisor-receptor. La clásica teoría monoaminérgica de la depresión ha sido reconsiderada, así como el papel de los cambios en la densidad de receptores noradrenérgicos y serotoninérgicos. En estos estudios se pone de manifiesto que el tratamiento prolongado con antidepresivos activa el sistema del AMPc y al cAMP Response Element Binding Protein (CREB), en ciertas regiones cerebrales.
La citada activación provoca la regulación de genes específicos, incluyendo un incremento en la expresión del Brain-Derived Neurotrophic Factor (BDNF) en hipocampo y en corteza cerebral. Como consecuencia de ello, se produce, probablemente, una respuesta terapéutica debida a la modificación de la actividad funcional de determinados circuitos cerebrales.
Por otra parte, la manipulación farmacológica del eje hipotálamo-hipófiso-adrenal (HHA) parece muy prometedora en el desarrollo de nuevos agentes antidepresivos. Finalmente, se revisan los mecanismos no aminérgicos que pueden tener interés en el conocimiento de las bases patológicas de la depresión, así como en el mecanismo de acción de los antidepresivos.
Aspectos neurobiológicos de la depresión y su relación con el futuro del tratamiento antidepresivo.
C. Alamo; F. López-Muñoz; B. Martín; P. García-García; E. Cuenca.
Departamento de Farmacología, Universidad de Alcalá, Madrid.
PALABRAS CLAVE: antidepresivos, Perspectivas, Bases biológicas, CRF, BDNF, NMDA
(KEYWORDS: Antidepressants, perspectives, Biological basis, CRF, BDNF, NMDA)
página 1
[otros artículos] [7/2/2002]
Resumen
Esta revisión estudia los diferentes mecanismos de acción antidepresiva, considerando los cambios bioquímicos implicados en la depresión y como éstos son modificados por el tratamiento antidepresivo, actuando más allá del nivel de la interacción entre neurotransmisor-receptor. La clásica teoría monoaminérgica de la depresión ha sido reconsiderada, así como el papel de los cambios en la densidad de receptores noradrenérgicos y serotoninérgicos. En estos estudios se pone de manifiesto que el tratamiento prolongado con antidepresivos activa el sistema del AMPc y al cAMP Response Element Binding Protein (CREB), en ciertas regiones cerebrales. La citada activación provoca la regulación de genes específicos, incluyendo un incremento en la expresión del Brain-Derived Neurotrophic Factor (BDNF) en hipocampo y en corteza cerebral.
Como consecuencia de ello, se produce, probablemente, una respuesta terapéutica debida a la modificación de la actividad funcional de determinados circuitos cerebrales.
Por otra parte, la manipulación farmacológica del eje hipotálamo-hipófiso-adrenal (HHA) parece muy prometedora en el desarrollo de nuevos agentes antidepresivos. Finalmente, se revisan los mecanismos no aminérgicos que pueden tener interés en el conocimiento de las bases patológicas de la depresión, así como en el mecanismo de acción de los antidepresivos.
Abstract
This review considers the various modes of action of the different types of antidepressants. The biochemical changes that may be, causally to depression and the mechanisms whereby these changes may be attenuated by antidepressant treatment are considered. The monoamine theory of depression and changes in densities of noradrenergic and serotonergic receptors has been reconsidered. Recent studies have begun to characterize the actions of antidepressant treatments beyond the neurotransmitter and receptor level. These studies show that long term antidepressant treatments result in the sustained activation of the cAMP system and cAMP Response Element Binding Protein (CREB) in specific brain regions.
This activation leads to the regulation of specific target genes, including the increased expresion of Brain-Derived Neurotrophic Factor (BDNF) in the hippocampus and cerebral cortex. The resulting states likely produce therapeutic response by altering the funtional activity of certain neural circuits in the brain. On the other field, the pharmacological manipulation of hypothalamic-pituitary-adrenal (HPA) axis function in depression appears to have considerable promise for the development of novel treatment approaches. This review ends with a consideration of the nonaminergic changes that may be of interest in the pathologic basis of depression and in the mechanism of action of the antidepressants.
Introducción
El abordaje farmacoterapéutico de los trastornos afectivos, desde la perspectiva de la farmacología científica actual, tiene su origen en la década de los 50, la misma década en la que vieron la luz los primeros agentes antipsicóticos y ansiolíticos. Los grandes avances históricos en el tratamiento de los trastornos del humor pueden concentrarse fundamentalmente en el descubrimiento y utilización terapéutica de los antidepresivos tricíclicos (ADT) y de los inhibidores de la monoaminooxidasa (IMAO) en la década de los 50 y en la aplicación definitiva de las sales de litio en el tratamiento y la profilaxis de los trastornos del humor, durante la década de los 60.
La introducción de los denominados antidepresivos atípicos, heterocíclicos o de “segunda generación” (maprotilina, nomifensina, trazodona, mianserina, entre otros), durante la década de los 70, no supuso, en contra de lo que en su momento se postuló, avances trascendentales, ni desde el punto de vista terapéutico ni desde su perfil de seguridad, salvo en casos individuales, con respecto a los antidepresivos clásicos. Sin embargo, desde finales de los 80, la incorporación al arsenal antidepresivo de una serie de nuevas familias de fármacos ha aportado un avance terapéutico, en algunos casos y en otros una decepción, en el tratamiento de la depresión. Como eventos positivos entre estos nuevos antidepresivos podemos considerar, sobre todo, a los inhibidores selectivos de la recaptación de serotonina (isrs), así como a los inhibidores de la recaptación de noradrenalina y serotonina (IRNS), los antidepresivos noradrenérgicos y serotoninérgicos específicos (NaSSA) y el inhibidor selectivo de la recaptación de noradrenalina (ISRN) representado, hasta el momento, tan sólo por la reboxetina. Por el contrario, la aceptación de agentes que combinan la inhibición de la recaptación de serotonina con el bloqueo de receptores postsinápticos 5-HT2 (nefazodona) y la de los inhibidores selectivos y reversibles de la monoaminooxidasa (RIMA) no ha sido la deseada. La tabla I recoge a las distintas familias de fármacos antidepresivos, con su fármaco prototipo, relacionándolos con sus respectivos mecanismos de acción (Cuenca y cols. , 2000)
La introducción de los ADT inauguró una nueva era en el tratamiento de la depresión y siguen siendo, en la actualidad, agentes de referencia, tanto en investigación clínica, donde la imipramina sigue siendo el control positivo de eficacia antidepresiva en múltiples estudios clínicos comparativos con nuevos agentes, como en terapia antidepesiva donde mantienen las mismas tasas de eficacia que el resto de antidepresivos aparecidos posteriormente. Su principal inconveniente estriba en su elevada incidencia de efectos adversos, debido a su inespecificidad farmacodinámica y a su falta de “limpieza bioquímica” ya que bloquean múltiples tipos de receptores. Mientras los antidepresivos tricíclicos continúan utilizándose en clínica de una forma importante, los IMAO han sufrido una reducción en su utilización muy amplia debido, en gran medida, a sus problemas de interacciones con alimentos ricos en tiramina y con otros fármacos psicoestimulantes, que pueden desembocar en trágicas crisis hipertensivas. No obstante, son fármacos que deben permanecer en el armamentarium antidepresivo, cosa que, incomprensiblemente, no sucede en España, ya que las depresiones atípicas, entre otras, siguen siendo candidatas a ser tratadas con estos fármacos y los RIMAS han sido una experiencia, en general, decepcionante.
La introducción en clínica, desde finales de los 80, de una nueva serie de familias de fármacos, entre los que destacan, por su gran utilización y aceptación, los ISRS que aportaron, frente a sus predecesores, considerables ventajas, sobre todo desde la perspectiva de la tolerabilidad general, seguridad en sobredosis y perfil de efectos adversos diferencial, lo que permitió abrir el campo de la terapéutica antidepresiva a facultativos no psiquiatras. En realidad, estos agentes han facilitado el tratamiento de la depresión en atención Primaria basado, más que en una diferenciación importante desde el punto de vista de eficacia terapéutica, en una mejor aceptación de la medicación por parte de los pacientes, lo que facilita la cumplimentación de la terapéutica, evita las recaídas y, además, proporciona una mejoría en su calidad de vida y han incrementado su espectro de indicaciones más allá de las propiamente antidepresivas (Alamo y cols. , 2000).
Pese a la importancia terapéutica de los psicofármacos, todavía se sigue haciendo una terapéutica fundamentalmente empírica que, no obstante, ha permitido, en gran medida, ir conociendo algunas bases de las enfermedades mentales y de su tratamiento. Pese a ello, Dentro del terreno de los antidepresivos, los mecanismos que se han explorado, durante estos cuarenta años, se centran fundamentalmente en el campo de las monoaminas. Quizá éste sea el hecho que condiciona una eficacia antidepresiva similar, alrededor del 70% de los pacientes, y que determina, para todos ellos, un inicio de acción lento, generalmente superior a las dos semanas.
Durante estas décadas se ha intentado explicar como actúan los antidepresivos, hecho que se ha visto dificultado con la observación de que se consiguen efectos terapéuticos, relativamente homogéneos desde el punto de vista porcentual, entre agentes que actúan a través de mecanismos diferentes e incluso opuestos. La explicación monoaminérgica de la depresión y del tratamiento antidepresivo está sometida a múltiples contradicciones (lapsus en el inicio de acción, eficacia relativa, acción inhibidora de la recaptación de monoaminas por la mayoría de antidepresivos frente la potenciadora observada con el caso de la tianeptina) e intentar explicar que un antidepresivo actúa porque incrementa en la hendidura sináptica la tasa de monoaminas (serotonina y/o noradrenalina) parece tan simple como intentar centrar el “mecanismo de acción” de un ordenador en su interruptor. Recientemente se han elaborado nuevas teorías que intentan explicar, no sólo como actúan los antidepresivos, sino también porqué son eficaces las medidas terapéuticas en un determinado porcentaje de pacientes y en otros no, así como cuales son las causas de los distintos tipos de depresión.
De la farmacología sináptica a la intracelular en la investigación de la depresión y su terapéutica
Como anteriormente hemos señalado, durante las décadas de los sesenta y setenta, la mayoría de los estudios, encaminados a demostrar los efectos de los psicofármacos sobre el sistema nervioso central (SNC) estaban centrados en aspectos extracelulares de la transmisión sináptica (Fig. 1), implicando, fundamentalmente, a la interacción del neurotransmisor con su receptor. Esta interacción era consecuencia de una acción sobre la inhibición de los sistemas de recaptación (ADTs) y de metabolización (IMAOs), que incrementaba los niveles de monoaminas en la hendidura sináptica lo que facilitaba su acción sobre el receptor.
En los últimos treinta años, la hipótesis monoaminérgica de la depresión ha marcado el desarrollo de nuevos agentes antidepresivos. En síntesis, esta hipótesis se basa en la capacidad de la reserpina, que como sabemos produce una depleción de monoaminas, de inducir cuadros depresivos en algunos sujetos (Schildkraut, 1965; Bunney y Davis, 1965; Coppen, 1967). El hecho de que los antidepresivos, hasta el momento conocidos, provoquen un incremento de monoaminas en la hendidura sináptica apoya la hipótesis de una disminución de la neurotransmisión monoaminérgica en la patogénesis de la depresión. Basándonos en los hechos comentados, los mecanismos hasta ahora explorados, para el hallazgo de nuevos antidepresivos, se han centrado, casi exclusivamente, en provocar un incremento de monoaminas en la hendidura sináptica. Estas modificaciones bioquímicas ocurren de forma rápida y pueden ser detectadas tras la primera dosis del fármaco antidepresivo. Sin embargo, el efecto terapéutico no se produce hasta pasadas algunas semanas de tratamiento, lo que parece indicar que el citado efecto se produce tras una serie de adaptaciones a nivel neuronal, como consecuencia de la administración crónica de estos agentes.
Este hecho dio paso, en la década de los ochenta, a la teoría de la adaptación receptorial. Según esta teoría, la activación persistente de receptores, como consecuencia de la elevación de serotonina y noradrenalina en la hendidura sináptica, llevaba a los mismos (5HT2 y b-adrenérgicos) a una hiporegulación (“down regulation”), fenómeno coincidente en el tiempo con el inicio del efecto terapéutico del antidepresivo (Sulser y cols. , 1978). Sin embargo, el hecho de que este fenómeno regulador no sea universal para todos los antidepresivos y que, por otra parte, los bloqueantes de receptores b adrenérgicos carezcan de efecto antidepresivo, e incluso puedan inducir depresión en algunos sujetos (Paykel y cols. , 1982), cuestiona la posibilidad de que este mecanismo adaptativo receptorial sea el único responsable del efecto terapéutico.
Como consecuencia de lo expuesto, comienza a existir una mayor apreciación de la complejidad en la transmisión sináptica, aceptándose que la regulación de la unión del neurotransmisor con el receptor representa sólo una pequeña parte de los efectos de los neurotransmisores sobre sus neuronas dianas, comportándose como un interruptor del mecanismo bioquímico responsable del efecto antidepresivo. En realidad, va quedando cada vez más claro que los neurotransmisores regulan todos los procesos que ocurren dentro de las neuronas. Además, durante esta década se ha reconocido que los efectos de los neurotransmisores sobre las neuronas diana no son directos, sino que se producen a través de cascadas bioquímicas de mensajeros intracelulares. Entre estos mensajeros intracelulares se encuentran proteínas que están unidas a las membranas, como las proteínas G (PG), segundos mensajeros como el AMP cíclico (AMPc) o como el propio calcio intracelular y proteínas fosforilizadoras que, modificando la dotación de fosfatos de todos los tipos de proteínas neuronales, alteran su función, siendo los responsables del amplio espectro de respuestas biológicas que se producen en la neurona, incluyendo las modificaciones en la expresión génica de las mismas (Fig. 1).
En los años 90, Blier y de Montigny (1994) involucran al receptor 5HT1A en el mecanismo responsable común de la actividad antidepresiva. Según estos autores, los distintos grupos de antidepresivos, incluyendo el electrochoque (ECT), a través de diferentes mecanismos, incrementarían la transmisión serotoninérgica a nivel del hipocampo. Así, se sabe que el retraso en el inicio de la acción de los inhibidores selectivos de la recaptación de serotonina (isrs) es debido al tiempo necesario para que se desensibilicen los autorreceptores 5-HT1A, presentes en el cuerpo celular de las neuronas serotoninérgicas. La desensibilización de estos autorreceptores permite la potenciación de una neurotransmisión serotoninérgica postsináptica. En la actualidad, se piensa que este mecanismo puede ser necesario, pero insuficiente, para explicar el efecto antidepresivo y que la intervención de factores adicionales debe ser tomada en consideración (Duman y cols. , 1997).
Además, los antidepresivos son capaces de regular otros subtipos de receptores de monoaminas hecho que, más que relacionarse con el efecto terapéutico, parece un correlato bioquímico secundario al aumento de monoaminas en la hendidura sináptica y/o al bloqueo receptorial que, además, provocan. Entre las adaptaciones observadas de manera más consistente, en la corteza cerebral de rata, tras tratamiento crónico con antidepresivos, se encuentra la regulación a la baja de receptores postsinápticos (a2, 5HT2, b). No obstante, es importante enfatizar que, en la actualidad, no existen evidencias convincentes de que la regulación de los receptores adrenérgicos o serotoninérgicos “per se” sea la responsable única del efecto terapéutico de los antidepresivos. Por tanto, mejor que llegar al concepto de que la regulación de receptores sea el mecanismo responsable de la acción de los agentes antidepresivos, parece más plausible que sea interpretado como marcador de un mecanismo de adaptación más complejo.
De esta forma, en la década de los noventa, se ha llegado a la consideración de que los mecanismos de transmisión sinápticos son aún más complejos. En la actualidad, se conoce que la regulación de la unión neurotransmisor con el receptor y los procesos de segundos mensajeros comentados forman sólo una pequeña parte de los mecanismos responsables de la respuesta neuronal. Además, las neuronas poseen una gran cantidad de proteínas tirosin-kinasas incrustadas en la membrana celular, que sirven de receptores para neurotrofinas y otros factores de crecimiento (Fig. 1).
En el contexto de los conocimientos comentados, la información más reciente da a entender que muchos agentes psicotrópicos interactúan inicialmente con proteínas extracelulares, denominadas receptores, a nivel sináptico, que, a través de mensajeros intracelulares, son los responsables de numerosas acciones de estos fármacos. Además, estos mensajeros intracelulares juegan un papel central en la mediación de los efectos a largo plazo que estos fármacos ejercen sobre la función cerebral, gracias a cambios neuronales fenotípicos, como la regulación a la baja, (“down regulation”), de receptores, síntesis de proteínas, liberación de neurotransmisores, etc. , consecuencia lógica de modificaciones en la expresión génica. Estos mecanismos adaptativos provocados por los antidepresivos son fundamentales para poder conocer nuevas formas de actuación en este terreno. Actualmente, estudios de biología celular y molecular han dado paso al conocimiento de modificaciones en los sistemas de transducción intracelular y en la regulación de la expresión de genes específicos producidos como consecuencia de la actuación sostenida de los antidepresivos. Hemos pasado, por tanto, de una psicofarmacología superficial y sináptica, a lo que podríamos denominar una psicofarmacología intracelular, que explicaría algunos de los datos hallados en décadas anteriores (Fig. 1).
En la actualidad, un objetivo prioritario para la neuropsicofarmacología es descubrir, con precisión, la naturaleza y los circuitos responsables de las modificaciones del funcionamiento neuronal que llevan a la depresión, así como los mecanismos de adaptación que ponen en marcha los antidepresivos para corregir y normalizar las alteraciones conductuales, cognitivas, afectivas y neurovegetativas observadas en estos cuadros. Para ello, la investigación no debe limitarse a los mecanismos aminérgicos clásicos (recaptación y metabolización) o más modernos (mecanismos receptoriales), sino que debe explorar otros conocimientos que la bioquímica, la biología molecular, la genómica, están constantemente aportando. Algunos de los conocimientos más recientes sobre estos mecanismos, se exponen a continuación.
Influencia de los antidepresivos con las vías de transducción intracelulares acopladas a receptores controladas por segundos mensajeros
En general, las vías de transducción de la señal intracelular se pueden dividir en dos categorías (Fig. 2):
Una categoría incluye vías que son controladas por segundos mensajeros acoplados al receptor. A modo de ejemplo, podemos citar al AMPc, GMPc, inositol trifosfato, calcio divalente (Ca++) y óxido nítrico (NO), que son usualmente regulados por neurotransmisores clásicos, como las monoaminas, aminoácidos, hormonas y neuropéptidos.
Una segunda categoría incluye vías que son controladas por receptores que interactúan con proteín-tirosin-kinasas y que son usualmente regulados por factores neurotróficos y citokinas. La activación de estos receptores da lugar a la regulación de una serie de cascadas intracelulares, como por ejemplo la MAP-kinasa o la denominada jak-STAT.
La regulación de estas dos grandes categorías de mecanismos de transducción intracelulares provoca cambios, a corto y largo plazo, responsables, en definitiva, de la función neuronal. Alteraciones en cualquiera de los múltiples mecanismos integrantes de las mismas pueden traducirse por cuadros psicopatológicos, que serían susceptibles de ser corregidos con la reconducción de los mismos a la normalidad. Además, en la actualidad se conocen otra serie de elementos, como por ejemplo diversos factores de transcripción, etc. , que forman parte de las mismas y que nos permiten su estudio en condiciones fisiológicas, en modelos fisiopatológicos, e, incluso, en los mismos pacientes, así como observar las modificaciones de los distintos escalones bajo tratamiento con antidepresivos y con otros agentes psicotrópicos, lo cual constituye, sin duda, un enorme avance para el conocimiento de lo que podría ser el inicio de la psicofarmacología del próximo milenio.
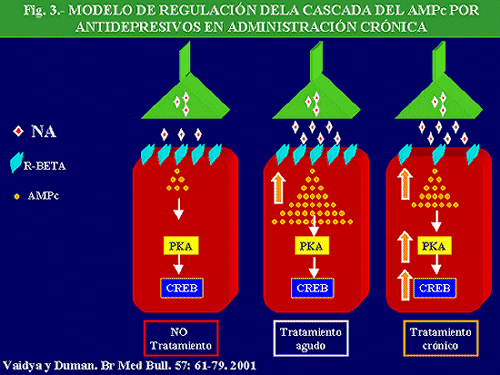
Entre los sistemas de transducción influenciados por los antidepresivos la cascada del AMPc es de los primeros que han sido señalados. En este sentido, pueden considerarse clásicos, los estudios que ponen de manifiesto que la producción de AMPc tras estimulación de receptores b es hiporegulada por la administración prolongada de antidepresivos (Fig. 3).
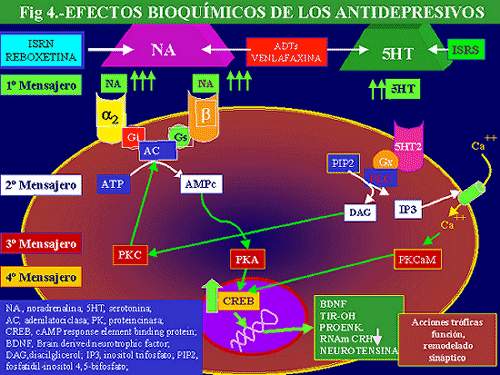
En la vía intracelular comentada, el papel de las proteínas G (PG) en la modulación de señales receptoriales ha tenido, durante los últimos años, un gran interés. ello es debido a que estas proteínas constituyen el paso post-receptorial inicial, no sólo en la vía del AMPc sino también en las más importantes vías de transducción intracelular. Las acciones de los receptores ligados a las PG están mediadas por dos sistemas de segundos mensajeros: por un lado, el sistema de la adenilatociclasa (AC) y, por otro, el sistema del fosfoinositol. Los elementos resultantes de la activación de estos sistemas, el AMPc y el diacilglicerol (DAG), respectivamente, activan distintos sistemas de fosforilización: el AMPc activa a la proteín-kinasa A (PKA), mientras que el DAG activa a la proteín-kinasa C (PKC) (dependiente de calcio y fosfolípidos), así como la CaM-kinasa II (dependiente del calcio y caldmodulina) (Fig. 4).
Los antidepresivos modifican el funcionalismo de distintas fracciones de estas PG
A modo ilustrativo, podemos señalar que la administración crónica de amitriptilina o iprindol, así como la ECT, elevan el acoplamiento entre la subunidad GS, la proteína G estimuladora, a la unidad catalítica de la AC (Ozawa y cols. , 1991). Por otra parte, Lesch y cols. (1991) han demostrado que los ADTs, en administración crónica, disminuyen la expresión de la GS y de la subunidad a de la GI en cerebro de rata, especialmente en corteza cerebral. Estos autores han detectado cambios en la expresión del RNA mensajero de diversas unidades de proteínas G bajo tratamiento con fluoxetina; de esta forma, se ha podido observar una disminución en la subunidad a de la GS en cerebro medio y un incremento en la expresión del RNA mensajero de la proteína Gaq y Ga12 en núcleo neoestriado y corteza frontal.
Por otra parte, se ha comprobado que en ratones carentes de proteína Gz (Knockout) desaparece el efecto antidepresivo experimental, en el test de natación forzada, de la reboxetina y de la desipramina, antidepresivos ambos inhibidores de la recaptación de noradrenalina. Este hecho parece indicar la necesidad de esta PGz para que se manifieste el efecto antidepresivo de estos agentes noradrenérgicos (Jing Yang y cols. , 2000).
Estos hallazgos han hecho pensar que en la depresión pueda existir algún trastorno en la superfamilia de PG acopladas a receptores y que los antidepresivos actuarían modificando, se supone que hacia la “normalidad”, esa disfunción de la PG. Así, se ha podido observar que la actividad de PG plaquetaria de pacientes deprimidos se encontraba disminuida respecto al grupo control. El tratamiento con antidepresivos, de forma continuada, normalizaba la actividad de esta PG en aquellos pacientes en los que se obtenía respuesta antidepresiva, mientras que sus niveles y actividad permanecía baja en los pacientes que, pese a estar tratados, no mejoraban de su cuadro depresivo (Karege y cols. , 1998). Igualmente se ha podido observar que los niveles de PGsa y de PGia en leucocitos de pacientes deprimidos fueron inferiores a los de individuos sanos. El tratamiento con ECT llevó a una normalización de estas PG (Avissar y cols. , 1998). Esta es una demostración, desde luego indirecta, ya que el estudio se ha realizado en células hemáticas, de una posible patología de las PG en la depresión y de cómo se normaliza en aquellos pacientes en los que el tratamiento antidepresivo resulta eficaz.
En este sentido, se ha hipotetizado que la base de estas alteraciones de las PG tuviera relación, en algunos casos, con un trastorno genético o con modificaciones producidas por distintas hormonas o por situaciones, como el estrés, que puedan alterar los mecanismos homeostáticos en los que intervienen algunas de estas hormonas. De hecho, distintos experimentos han demostrado que, tanto la subunidad a de la proteína GS, como la de la proteína GI están bajo control de los glucocorticoides. Así, se sabe que el litio y algunos antidepresivos cambian la expresión de los receptores tipo II de los glucocorticoides en cerebro de rata, así como la actividad de distintas subfracciones de las PG. Estos cambios podrían guardar alguna relación con las alteraciones del humor detectadas en ciertos periodos como la pubertad, el síndrome disfórico premenstrual, la menopausia, así como en diferentes eventos estresantes presentes a lo largo de la vida, en los que la influencia hormonal es la norma (Bourin y Baker, 1996).
Por todo ello, se ha pensado que un objetivo terapéutico en el abordaje de los trastornos afectivos podría implicar la actuación de agentes farmacológicos sobre las PG. En este sentido, la síntesis de fármacos que actúen de forma selectiva sobre subunidades de estas proteínas podrían facilitar el logro de los efectos deseados. Es concebible que la síntesis de estos futuros medicamentos pueda ser obtenida a partir de un mayor conocimiento de las interacciones específicas de las toxinas bacterianas y víricas sobre este sistema de PG. De momento, no disponemos de fármacos dentro de este perfil que sean lo suficientemente específicos como para obtener los efectos deseados, sin que provoquen efectos adversos importantes (Bourin y Baker, 1996). No obstante, suele ser un buen barómetro de la importancia de estos hallazgos el hecho de que investigadores de la Industria Farmacéutica estén desarrollando agentes capaces de modular de forma directa a estas PG ya sea estimulándolas, como sucede con el SCT-11 de Laboratorios Lilly, en la actualidad en fase clínica I, o inhibiéndola como es el caso del GPCR de Laboratorios Roche aún en fase de desarrollo preclínico (Pharma Projects 2001).
Como hemos señalado, la vía de transducción dependiente del AMPc puede ser regulada, tras la administración de antidepresivos, por la estimulación tanto de receptores serotoninérgicos como noradrenérgicos. Así, en un paso intracelular más allá del desarrollado con las PG, se sabe que la administración prolongada, aunque no aguda, de antidepresivos puede modificar los sistemas de fosforilización AMPc dependiente en áreas cerebrales concretas. Esto se realiza mediante la activación de proteín-kinasas, especialmente la PKA, que regulan factores de transcripción que influyen en la expresión de proteínas específicas adicionales (Brunello y Racagni, 1998).
En la línea de lo comentado, se ha podido constatar que la administración crónica de una gran variedad de antidepresivos disminuye la producción de AMPc, estimulada por foskolina, en hipocampo (Newman y cols. 1993), fenómeno secundario, en la mayoría de los casos, a una disminución de la densidad y función de receptores b adrenérgicos que, como sabemos, están acoplados a esta vía transduccional. En este sentido, hemos de señalar que diversos agentes antidepresivos, como imipramina o tranilcipromina, en administración prolongada, o la ECT, aumentan la actividad de la PKA no soluble y la disminuyen en la fracción citosólica de la corteza frontal (Nestler y cols. , 1989). Brunello y cols. (1990) obtuvieron un incremento de la subunidades reguladoras de la PKA en fracción soluble de corteza frontal de rata, tras la administración crónica de desmetilimipramina (DMI). Además, Pérez y cols. (1994) hallaron que la administración crónica de fluoxetina o de DMI incrementó la unión del AMPc a la subunidad regulatoria de la PKA.
Asimismo, se ha podido detectar que algunos agentes tienen comportamientos diferenciales sobre los sistemas de transducción AMPc dependientes y sobre sistemas de PKs dependientes de calcio y caldmodulina. La PKC es una enzima clave en la fosforilización de proteínas celulares y su actividad ha sido asociada con la actividad intracelular secundaria a activación de segundos mensajeros, como calcio y DAG. En este sentido, es destacable que dos agentes antidepresivos, como fluoxetina y desipramina, que actúan a través de mecanismos serotoninérgicos y noradrenérgicos respectivamente, administrados de forma repetitiva, modifican, de forma similar, la actividad basal de la PKC, aunque presentan un comportamiento diferente sobre la PKC, tras estimulación del receptor 5HT2 por un agonista (DOI). Este hecho es indicativo de una desensibilización del receptor serotoninérgico, consecuente con la administración de fluoxetina, fenómeno no observado con DMI. En definitiva, estos resultados hablan de la manifestación de una adaptación post-receptorial, que implica vías de transducción independientes del AMPc, inducida por el tratamiento antidepresivo continuado (Mann y cols. , 1995).
Algunos de los pasos de las diferentes vías intracelulares que nos ocupan pueden ser modificados de forma diferencial por distintos tipos de antidepresivos (TABLA II). Así, el inhibidor selectivo de la recaptación de noradrenalina, reboxetina, no modifica la fosforilización de una proteína específica del microtúbulo (MAP2) AMPc dependiente, en contraste con lo que sucede con los ADTs y los ISRSs. De igual modo, mientras que los ADTs y los ISRSs son capaces de potenciar la unión del AMPc a la subunidad regulatoria de esta PK, la reboxetina hace todo lo opuesto. Por el contrario, el comportamiento de la reboxetina es idéntico al de los ISRS en lo referente al incremento de la actividad fosforilizadora de PK calcio y caldmodulina dependiente. Por consiguiente, pese a que los diferentes tipos de antidepresivos pueden compartir mecanismos centrales comunes, la vía por la que afectan los citados mecanismos puede ser diferente (Brunello y Racagni, 1998).
El CREB (cAMP Response Element Binding Protein), diana post-receptorial común para los antidepresivos
Existen algunos cambios bioquímicos comunes a la acción de los antidepresivos. Entre los más conocidos y constantes, ya que se producen con todos los agentes con los que este parámetro ha sido estudiado, se encuentran las modificaciones hipocampales, así como en otras estructuras cerebrales, del CREB. El CREB es un factor de transcripción encargado de mediar en gran parte de las acciones del sistema del AMPc sobre la expresión génica (Meyer y Habener, 1993; Ghosh y Greenberg, 1995), estimulando la producción de diversas proteínas entre las que se encuentran algunos factores neurotróficos, en especial el conocido con las siglas BDNF (Brain Derived Neurotrophic Factor), que será motivo de comentarios posteriormente.
Como ya se ha señalado la vía del AMPc, y por tanto el CREB, es potencialmente modificable por la serotonina y la noradrenalina, ya que está relacionada con subtipos de receptores para ambas (5HT4, 5HT6, 5HT7 y b adrenérgicos). Además, este sistema CREB puede ser considerado como diana intracelular para antidepresivos y otros agentes, a través de otros receptores de serotonina y noradrenalina (5HT2A, 5HT2C y a1-adrenérgicos), que pueden activarse secundariamente a la estimulación de PKs Ca++ dependientes. De lo expuesto puede deducirse que el CREB está sometido a la acción de antidepresivos que son capaces de actuar potenciando la transmisión serotoninérgica y/o noradrenérgica, prácticamente todos los conocidos de momento, habida cuenta de que los interruptores que ponen en marcha las distintas cascadas que en definitiva llevan a una modificación de este factor de transcripción pueden ser tanto receptores noradrenérgicos como serotoninérgicos (Fig. 4).
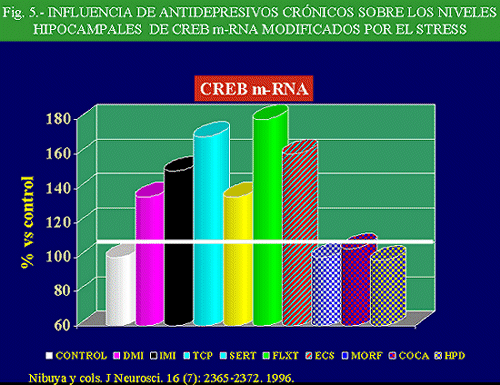
En este sentido, no es de extrañar que la regulación al alza del CREB se observe tras la administración de diversos antidepresivos, habiéndose podido demostrar que, tras tres semanas de tratamiento con fluoxetina, o diez días con ECT, aumenta la función y la expresión del CREB en hipocampo. Por el contrario la administración de otras substancias psicoactivas, como la cocaína, morfina o el antipsicótico haloperidol no alteraron los niveles de CREB (Nibuya y cols. , 1996) (Fig. 5).
A conclusiones similares llegan otros grupos de trabajo que ponen de manifiesto que la desipramina, inhibidor de recaptación de noradrenalina, la fluoxetina, de serotonina, o la tranilcipromina, inhibidor de la MAO, administrados de forma crónica a ratones transgénicos (CRE-lacZ), incrementan la actividad del CREB sobre la transcripción genética en diversas áreas límbicas e hipotálamo (Thome y cols. , 2000). Estos datos indican que los antidepresivos, independientemente del mecanismo sináptico inicialmente implicado, provocan una regulación al alza del CREB y que el tiempo necesario para que se produzca la inducción del CREB está cronológicamente relacionado con el efecto terapéutico del tratamiento antidepresivo.
Los datos anteriormente expuestos guardan una cierta analogía conceptual con los observados en estudios “postmorten” de cerebros de pacientes depresivos, en los que se ha podido demostrar que existe una disminución de los niveles de CREB en corteza temporal respecto a los controles (sujetos sin patología psiquiátrica, esquizofrénicos o maníacos sin tratamiento o bajo tratamiento antipsicótico). Por el contrario, las muestras obtenidas de sujetos depresivos sometidos a tratamiento exhibían una mayor tasa de CREB. Este incremento, similar a lo comentado en animales de experimentación, fue selectivo, no se manifestó en otras áreas corticales estudiadas, como la occipital, ni en muestras de individuos tratados con otros agentes psicofarmacológicos no antidepresivos (Dowlatshahi y cols. , 1998). De todo lo expuesto, parece poder postularse que en la depresión existe un déficit funcional centrado en el CREB, punto de encuentro de vías noradrenérgicas y serotoninérgicas, que es incrementado bajo tratamiento con diferentes tipos de antidepresivos (fig. 4).
Existen evidencias adicionales de que el CREB juega un importante papel en el mecanismo de acción antidepresivo. En efecto, se puede incrementar la vía funcional del AMPc a través de una inhibición de la fosfodiesterasa, enzima encargada de metabolizarle. Existen distintas familias de fosfodiesterasas siendo la más selectiva del AMPc la PDE4. El rolipram, un inhibidor de la PDE4, exhibe propiedades antidepresivas en modelos experimentales en roedores, así como en algunos estudios realizados en pacientes depresivos. Los efectos adversos, en especial las nauseas, han hecho que el rolipram no sea una realidad terapéutica. No obstante, se realizan estudios para identificar subtipos de PDE4 que no estén expresados en áreas relacionadas con el vómito (postrema) y que se localicen de forma selectiva en áreas límbicas, lo que constituiría una buena diana terapéutica para nuevos antidepresivos, así como para agentes potenciadores de la respuesta de otros antidepresivos (Duman, 2000). En cualquier caso, los datos comentados ponen de manifiesto el papel del CREB como elemento clave en el mecanismo de acción de los antidepresivos.
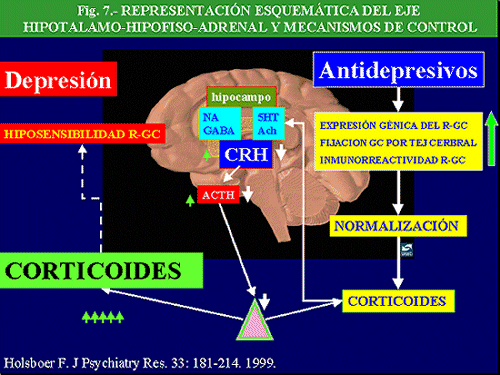
Por otra parte, el CREB regula el gen c-fos, responsable de la proteína c-fos, que a su vez es uno de los componentes del factor de transcripción AP-1 (Proteína activadora 1). La importancia de ésta AP-1 viene dada por su capacidad de regular la expresión génica de la tirosina-hidroxilasa, paso limitante en la síntesis de catecolaminas, de la proencefalina y de la neurotensina. De igual manera el incremento de CREB disminuye la expresión del RNAm de la CRH, hormona que como veremos se encuentra elevada en, al menos, algunos tipos de depresión (Fig. 4) (Dahmen y cols. , 1997).
En un elegante estudio, realizado recientemente por el grupo de Dumas (Chen y cols. , 2001), se ha demostrado que la administración de CREB, usando como vector el virus del herpes, en hipocampo produce un efecto antidepresivo experimental tanto en el test de inmovilización como en el de indefensión aprendida. Por el contrario, los animales que recibieron, como controles, sól el virus del herpes sin CREB, no mostraron respuesta antidepresiva. Estos datos sugieren que el efecto antidepresivo inducido por agentes que elevan los niveles de CREB en hipocampo (antidepresivos, ECT) puede estar mediado por esta sustancia y que el CREB constituye una diana molecular para el estudio de nuevos agentes antidepresivos.
Los fenómenos comentados respecto al CREB constituyen un apoyo sobre la participación de este sistema de transducción hipocampal como responsable de la regulación de una serie de genes específicos implicados en el efecto “normalizador” de los antidepresivos. Uno de los objetivos actuales es descubrir estos genes y determinar su relevancia en la respuesta clínica al tratamiento antidepresivo.
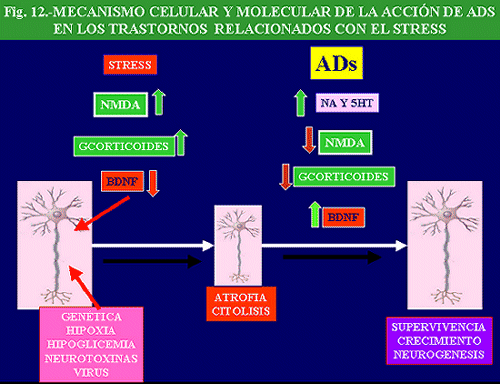
Papel de las neurotrofina (BDNF) en la depresión y su relación con el efecto antidepresivo
Como se ha comentado anteriormente, el CREB estimula la activación de genes responsables de la síntesis de diversas proteínas una de las cuales, BDNF (factor neurotrófico derivado del cerebro) parece jugar un importante papel en los mecanismos que rodean al fenómeno depresivo y al efecto de los antidepresivos. El BDNF fue descubierto por su papel en el desarrollo neural de animales jóvenes, pero en el adulto parece actuar protegiendo a las neuronas del estrés, a la vez que facilita la creación de nuevas conexiones. Como gráficamente señala Nestler “una neurona con más BDNF” es una neurona más saludable y más feliz”.
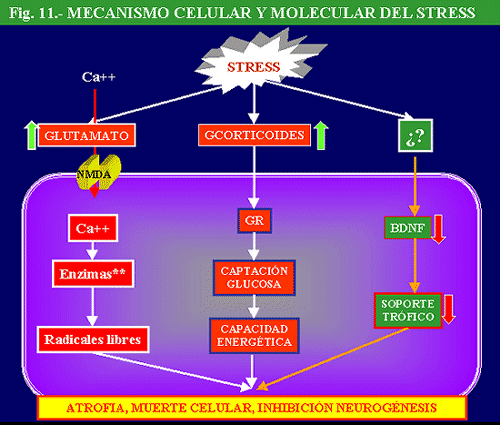
En esta línea, distintos tipos de neurotrofinas, así como sus correspondientes receptores, pueden constituir dos dianas de importancia en el desarrollo de conocimientos sobre los mecanismos básicos implicados en la depresión y en su posible tratamiento. Así, existen evidencias que indican que el estrés disminuye la expresión de algunas neurotrofinas en el hipocampo y en el cortex frontal, y que ello puede aumentar la vulnerabilidad de las neuronas a distintas formas de agresión. Por el contrario, los antidepresivos (IMAOs o inhibidores de la recaptación de noradrenalina y serotonina –IRNS-, así como la ECT) aumentan la expresión de este factor neurotrófico y previenen la disminución del mismo inducida por el estrés. Estos y otros tipos de mecanismos nuevos pueden ser explorados para estudiar los efectos terapéuticos a largo plazo de los antidepresivos.
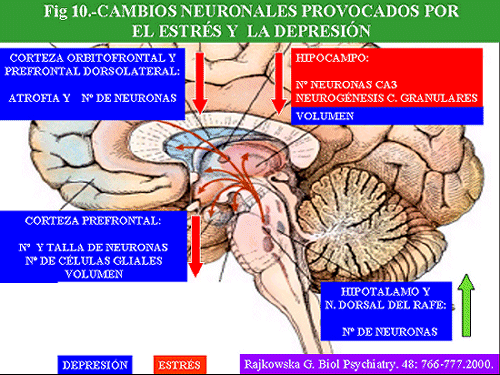
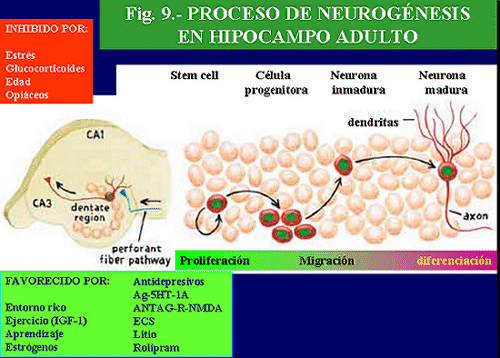
Clásicamente se ha considerado que el cerebro adulto era refractario a la regeneración y al nacimiento de nuevas neuronas. Sin embargo, estudios recientes han revelado que en diversas zonas cerebrales adultas, como en el girus dentatus del hipocampo y neocortex de mamíferos, incluidos los humanos, se produce el nacimiento de nuevas neuronas (neurogénesis) (Gould y cols. , 1999; Jacobs y cols. , 2000).
Entre los factores que pueden afectar la neurogénesis se encuentran las neurotrofinas. La familia de las neurotrofinas incluye, entre otras, la neurotrofina-3, neurotrofina-4 y también el factor neurotrófico derivado del cerebro (BDNF). Las acciones de las neurotrofinas son mediadas por la unión a receptores específicos, denominados trkB, que activan la proteín-tirosin-kinasa intracelular. Estos factores neurotróficos están implicados en la diferenciación y crecimiento de múltiples tipos de neuronas, durante el desarrollo cerebral, así como en el mantenimiento y en la supervivencia de neuronas y células no neuronales en el cerebro maduro. Así, se sabe que la supervivencia y el crecimiento de neuronas serotoninérgicas en el cerebro adulto es incrementada por el BDNF y, en menor grado, por la neurotrofina-3, pero no por el factor de crecimiento nervioso (NGF). Además, es conocido que las neurotrofinas pueden, en realidad, influenciar la función neuronal, como se demuestra por el hecho de que la exposición de cortes hipocampales al BDNF aumenta las conexiones de algunas neuronas a nivel sináptico. Por otra parte, las monoaminas son capaces de aumentar la acción neurotrófica, por lo que conjuntamente pueden participar en la regulación de las funciones cerebrales (Alamo y cols. , 1998).
Diversos hechos avalan la hipótesis de que las neurotrofinas puedan participar en el efecto terapéutico de algunos antidepresivos. Por una parte, se sabe que el BDNF exhibe propiedades antidepresivas en dos modelos experimentales de depresión, como el test de la natación forzada y el test de la indefensión aprendida (Siuciak y cols. , 1996).
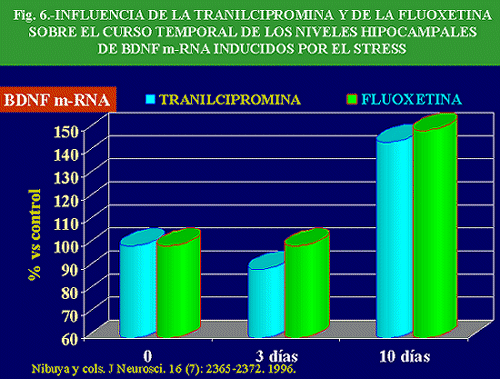
Además, la administración crónica, pero no aguda, de antidepresivos, entre los que se encuentran los IRNS, inhibidores selectivos de la recaptación de alguna de estas aminas, IMAOs y antidepresivos atípicos, así como la ECT, aumenta la expresión de BDNF (Fig. 6) y de los receptores de trkB en hipocampo, coincidiendo estos efectos temporalmente con el efecto terapéutico (Nibuya y cols. , 1996). Además, el efecto de los antidepresivos se potencia con un cierto grado de ejercicio físico en el animal de experimentación (Russo-Neustadt A y cols. , 2001).
El BDNF es capaz también de aumentar el crecimiento de neuronas noradrenérgicas y serotoninérgicas, y proteger a estas neuronas del efecto neurotóxico. Por tanto, estos hechos apoyan el papel del BNDF en las acciones de los antidepresivos (Duman y cols. , 1997; Altar, 1999; Alamo y cols. , 1998; Skolnick, 1999; Duman 2000).
Existen algunos datos que relacionan el papel del BDNF con el CREB. En primer lugar, existe una coincidencia cronológica entre la regulación al alza del CREB y del BDNF, regulándose ambos factores en las mismas poblaciones neuronales del hipocampo. En segundo lugar, se ha podido demostrar que la activación de PK activadas por AMPc o calcio aumentan la expresión de BDNF (Ghosh y cols. , 1994; Condorelli y cols. , 1994). Además, la administración de inhibidores de la fosfodiesterasa, que incrementa los niveles de CREB, aumenta los niveles de BDNF (Duman, 2000). Por ultimo, la disminución experimental de CREB en hipocampo disminuye los niveles de BDNF y el incremento que de este factor neurotrófico provocan los antidepresivos (Duman y cols. , 1995).
Además, en necrópsias de sujetos tratados con antidepresivos se observó un incremento de BDNF en diversas zonas de hipocampo en relación con los niveles de sujetos no tratados (Chen y cols. , 2001). Estos datos concuerdan con los incrementos de CREB observados en necrópsias de pacientes tratados con antidepresivos y ponen de manifiesto que el incremento de la BDNF es secundario al aumento de CREB inducido por el tratamiento antidepresivo, no sólo en animal de experimentación sino también en humanos (Dowlatshahi y cols. , 1998).
Desde una perspectiva terapéutica, la posibilidad de incrementar la expresión de BDNF por inhibición de la metabolización del AMPc ha sido evaluada, mediante la administración crónica de inhibidores de la fosfodiesterasa (rolipram, RO 20-1724) de forma prolongada. En estas condiciones, se observa un aumento de la expresión del CREB y del BDNF en el hipocampo de rata, hecho que no se produce tras la administración aguda (Fujimaki y cols. , 2000; Nibuya y cols. , 1996). Por otra parte, los datos expuestos permiten la búsqueda de análogos del BDNF capaces de actuar sobre el receptor trk-B y poner en marcha los mecanismos intracelulares, supuestamente responsables del efecto de esta neurotrofina.
Eje Hipotálamo-Hipofiso-Adrenal su papel en la depresión y en el mecanismo de acción de los antidepresivos
El descubrimiento, hace un par de décadas, del papel de la hormona liberadora de corticotropina (CRH) en el control de las reacciones del eje hipofiso-adrenal, así como de otras zonas del SNC, ante el estrés, ha revolucionado nuestro conocimiento de la neurobiología de la depresión y ha abierto interesantes perspectivas terapéuticas. La CRH, también conocida como CRF (Corticotropin Releasing Factor), es un péptido de 41 aminoácidos, que se encuentra en regiones hipotalámicas y extrahipotalámicas, que gobierna la liberación de la ACTH (corticotropina) hipofisaria, la cual, a su vez, controla la liberación de cortisol de la médula adrenal. Los niveles de cortisol son controlados, a través de un mecanismo de retroalimentación, por una serie de receptores situados a nivel hipofisario, hipotalámico, así como en zonas del hipocampo. El cortisol tiene múltiples funciones, entre las que se encuentra una potenciación de la respuesta fisiológica ante el estrés. Ante diversas situaciones estresantes se elevan los niveles de corticoides lo que facilita un incremento de energía y una situación de alerta, ésta última a través de la estimulación adrenérgica, necesarias para enfrentarse a la situación. Sin embargo, cuando el cortisol es secretado en exceso y de forma crónica aparecen una serie de secuelas biológicas, hipertensión arterial, diabetes, arteriosclerosis, inmunosupresión, osteoporósis y atrofia muscular (Sadek y Nemeroff, 2000).
En la depresión se ha descrito una hiperactividad del eje HHA (Hipotálamo-Hipofiso-Adrenal), manifestada por un incremento de niveles plasmáticos, urinarios y en líquido cefalorraquídeo (LCR) de cortisol, un aumento de la frecuencia, magnitud y duración de los episodios de secreción de cortisol y ACTH, resistencia a la supresión por dexametasona, aumento de los niveles de CRH en LCR y en cerebros de depresivos “post morten” (Mitchell, 1998) y disminución del control de retroalimentación negativo de glucocorticoides.
Resulta a
IMPORTANTE: Algunos textos de esta ficha pueden haber sido generados partir de PDf original, puede sufrir variaciones de maquetación/interlineado, y omitir imágenes/tablas.




Hipomanía inducida por sertralina. A propósito de un caso.
IRENE PÉREZ SAGASETA DE ILURDOZ et. al
Fecha Publicación: 20/05/2024
La psicosis como síntoma nuclear en los trastornos de personalidad
Pedro Gómez Pérez et. al
Fecha Publicación: 20/05/2024
El doble filo del tratamiento con antidepresivos
Julia Rodriguez Martin et. al
Fecha Publicación: 18/05/2023
Efecto de los antidepresivos sobre la inflamación
Oscar Martin Santiago et. al
Fecha Publicación: 20/05/2022
Analgesia con antidepresivos
Joana Gonçalves Cerejeira et. al
Fecha Publicación: 20/05/2022
Beneficios de los antidepresivos en la esfera sexual
Carolina Alario Ruiz et. al
Fecha Publicación: 20/05/2022